

Abstract
Erosive rheumatoid arthritis (RA) is accompanied by synovial tissue hyperplasia associated with the proliferation of transformed-appearing synovial lining cells. In the present study we have analysed the expression of the p53 tumour suppressor gene in the synovial pannus tissue from patients at various stages of the disease. We used a combination of polymerase chain reaction (PCR) and single-strand conformation polymorphism (SSCP) on DNA and reverse transcription, PCR and sequencing on cDNAs from synovial tissues or purified synovial cell populations of 24 RA and three osteoarthritis (OA) patients. We also studied p53 expression by immunohistochemical analysis. Mutations suspected after SSCP were identified by systematic sequencing of the p53 exon 6, especially in the fibroblast-like, adherent synovial cell population, associated with an erosive disease. Some accumulation of the protein was detected in immunohistochemical analysis of the p53 tumour suppressor gene in the patients' synovial tissues. However, no sign of malignancy was seen in these patients after a 2-year survey. These results show some abnormalities in the p53 tumour suppressor gene in RA patients, but do not allow this to be related to characteristic proliferative features of the rheumatoid synovium.
Keywords: apoptosis, DNA damage, suppressor mutation, oncogenes, histocytochemistry
INTRODUCTION
Rheumatoid arthritis (RA) is a disease of unknown aetiology which involves excessive growth of the synovial membrane, leading to destruction of cartilage and bone. The molecular and cellular basis of rheumatoid joint destruction is characterized by abnormal expression of oncogenes modulating cellular proliferation and induction of lysosomal enzymes and metalloproteinases (reviewed in [1]). Cultured RA synovial cells display malignant cell features, e.g. a tendency to grow in disorganized monolayers and form foci, ability to form colonies in soft agar, presence of many multinucleated cells, and tumorigenicity in nude mice [1].
Several genes, particularly oncogenes [2] and tumour suppressor genes [3], can be involved in the regulation of the cell cycle process. Oncogenes are known to be able to stimulate and promote cell division, whereas tumour suppressor genes act by repressing cell proliferation. Inactivation of the p53 tumour suppressor gene by point mutations and/or allelic loss is the most frequent genomic alteration in human cancers [4]. The p53 protein is a sequence-specific DNA-binding protein that is active as a transcription factor, and can inhibit SV40 replication in vivo and in vitro (reviewed in [4]). The p53 protein was recently implicated in the induction of apoptosis [5], and activation of several genes, e.g. the mdr1 [6], gadd45, and mdm2 genes [4].
Immunohistological studies on normal cells showed that p53 levels were extremely low due to the very short half-life of the protein [7]. In cells exposed to ionizing radiation, ultraviolet light, or mitomycin C, or in those infected with certain DNA viruses, there is a very high p53 accumulation [8], which occurs secondarily to half-life prolongation of the mutated protein due to p53 gene alterations [7]. Clones of p53-mutated keratinocytes frequently accumulate in normal human skin, and can be expanded after sunlight exposure [9]. The p53 response to ionizing UV or x-ray radiation is defective in patients suffering from benign conditions such as ataxia telangiectasia, or xeroderma pigmentosum, respectively [8]. In psoriasis, an autoimmune disease, abnormal nuclear p53 immunohistochemical staining in the basal layer of psoriatic skin lesions has been reported [10], suggesting specific genetic alterations, but this was not confirmed using polymerase chain reaction (PCR)-single-strand conformation polymorphism (SSCP) and immunohistological analysis [11]. Finally, frequently elevated serum levels of antibodies against p53 are frequently detected in patients with systemic autoimmune diseases such as systemic lupus erythematosus (SLE) [12].
In the present study, we looked for mutations of the p53 gene in synovial membrane and purified adherent synoviocytes of a series of 24 RA and three osteoarthritis (OA) patients by a combination of SSCP, reverse transcription (RT), PCR, dideoxy-sequencing and p53 expression in immunohistochemical analysis.
MATERIALS AND METHODS
Patients and tissue samples
Synovial membranes were obtained from 24 patients diagnosed as having RA (Table 1), according to the American College of Rheumatology (formerly the American Rheumatism Association) 1987 revised criteria [13], and three OA patients. After surgical excision, mainly for wrist synovectomy, all samples were cut in small pieces, rapidly frozen in liquid nitrogen and subsequently stored at −80°C until use. Peripheral blood from each patient was taken in parallel, and mononuclear cells (PBMC) were separated over leucocyte separation medium (MSL, Eurobio, Les Ulis, France). Adherent cells were prepared by cultivating very small tissue pieces in culture plates and passaging once in culture flasks, leading mainly to type-B, fibroblast-like synoviocytes. Genomic DNA was extracted from PBMC, cultured cells and minced specimen pieces (≈ 100 mg) using the classical phenol/chloroform method, and total RNA was prepared by homogenization of small tissue pieces in ready-made RNA-Plus extraction solution (Bioprobe Systems, Montreuil, France), extracted by chloroform, precipitated by isopropanol and washed with ethanol.
Table 1.
Subjects studied

PCR-SSCP analysis
Radioactive PCRs were performed for 30 cycles for exons 5–1, 5–2, 7, 8 and 9, and 35 cycles for exons 2 and 6, consisting of 30 s at 94°C, 2 min at 65°C, and 2 min and 30 s at 72°C in a final volume of 25 μl, using 32P-dCTP as a label. Primer pairs for p53 gene analysis were from ref. [11] for exons 2, 5–1, 5–2, 7, and ref. [14] for exon 6. Screened regions covered exon 2 and exons 5–9. SSCP analysis was performed using a modified version of the method originally described by Orita et al. [15]. Briefly, PCR products were diluted first 1:10 in 0.1% NaDodSO4–10 mmol/l EDTA and then 1:2 in a sequencing stop solution containing 95% formamide, 0.025% bromophenol blue and 0.025% xylene cyanol. Samples were heated at 95°C for 5 min, chilled on ice, and immediately loaded (4 μl) onto a 6% non-denaturing acrylamide-bis-acrylamide-TBE gel in the presence or absence of 5% glycerol. Gels were run using TBE as a buffer, simultaneously at × 0.5 and × 1, at constant power and at room temperature for those containing glycerol, or at 4°C for those without glycerol. After migration, gels were subjected to autoradiography against a XAR film (Kodak, Rochester, NY) at −80°C.
cDNA sequencing
One microgram of each RNA from either RA or OA patients was reverse-transcribed with oligo-dT primer and Murine Moloney Leukaemia Virus reverse transcriptase according to the Superscript II kit recommendations (Life Technologies, Cergy-Pontoise, France). After amplification in the same conditions as for DNA, 5 μl of exon 6 PCR product for each cDNA were subjected to dideoxy sequencing with Sequenase version 2 system (Amersham, Paris, France) using the prescribed protocol. Briefly, PCR product was purified from excess primers and dNTPs with endonuclease and alkaline phosphatase for 30 min at 37°C, and enzymes were deactivated for 15 min at 80°C. Hybridization of the denatured template to the sequencing primer was done by heating at 100°C for 2 min in the thermal cycler and cooling immediately in an ice-water bath for at least 5 min. Sequence labelling was performed in a 35S-dATP-containing mix for 5 min at room temperature, and termination reactions were completed in prewarmed termination tubes in the thermal cycler for 10 min at 37°C before stopping reactions with stop solution. Samples were run on a 8% acrylamide-bisacrylamide sequencing gel containing 7 m urea in 1× TBE buffer. After fixation of the gel for 30 min with a 10% methanol–10% acetic acid solution, it was vacuum and heat dried, then exposed on a β-max Kodak film for 24–48 h.
Immunohistochemical analysis
Three different anti-p53 antisera were used: polyclonal antibody CM1 (Novocastra, Newcastle-upon-Tyne, UK), MoAbs DO7 (Dako, Versailles, France) and 1801 (Oncogene Science, Uniondale, NY). All of these antibodies recognized both the wild-type and mutated form of the p53 protein. The epitopes of both D07 and 1801 MoAbs were located in the NH2-terminal portion of the protein, and CM1 polyclonal antibody was raised against a recombinant wild-type p53 protein.
Formalin-fixed, paraffin-embedded tissue sections of the surgery specimen (5 μm thick) were cut and fixed in alcohol–acetone for 10 min at 4°C. Enzyme immunohistology was performed using the alkaline phosphatase technique [16]. Briefly, a 10 μg/ml MoAb dilution was applied to each slide and incubated for 40 min at 4°C. After washing, a 1:15 dilution of rabbit anti-mouse immunoglobulins (Dako) was added and incubated for a further 30 min, followed by addition of a 1:15 dilution of the alkaline phosphatase complex (Dako) and incubated for another 30 min. The stain was revealed with alkaline phosphatase substrate and fast-red as colouring agent, and counterstained with haematoxylin.
RESULTS
PCR-SSCP analysis of the p53 tumour suppressor gene
After PCR amplification of various portions of conserved domains of the p53 gene, e.g. exon 2, and exons 5–9, we tried to detect point mutations using the SSCP technique, based on conformation of the single-strand DNA [15]. Under non-denaturing electrophoretic conditions, migration of single-strand DNA is a function of its conformation, which depends on its sequence. Even single-point mutations can be detected by variant migration patterns or by the presence of additional bands, provided that the temperature and TBE buffer concentration in which gels are run are suitable [15]. Here, several gel migration conditions (e.g. TBE buffer × 0.5 or × 1; polyacrylamide gel with and without 5% glycerol; gel migration at 4°C or at room temperature) were used for each exon to improve sensitivity. Furthermore, given the fact that p53 tumour suppressor gene mutations have been associated with progression towards more aggressive cancer or disease [4], we studied many RA patients with chronic and severe forms of the disease, as shown in Table 1. As a positive control, we used previously sequenced, mutated DNAs from tumour cell lines which presented a shifted band (for XG5 cell line, normal bands absent for exon 8, [17]) or an additional band (for Daudi and Jurkat cell lines, exon 6, [18]), thus indicating point mutations. By comparison with a negative control DNA from normal or patient blood, identical wild-type migration patterns were observed in all RA for most of the exons, as found for exon 8, where normal bands were absent. However, for exon 6, faint abnormal patterns could not be excluded for samples from patients 1, 3, 5, 6 and 8, with an additional band comparable to the one of positive controls. We therefore decided to sequence exon 6 in all samples.
P53 exon 6 sequencing
PCR-amplified exon 6 was subjected to dideoxy-sequencing of single-stranded DNA from each RA patient, after validating the sequencing procedure by detecting point mutations for Daudi and Jurkat DNAs. Surprisingly, if most of the patients presented wild-type p53 exon 6, three out of 20 patients showed additional bands for that exon, evidencing point mutations. Figure 1 depicts the mutation of each of these patients compared with the wild type, either from normal or patient blood. The intensity of these bands clearly shows that the mutation is present only in a small subpopulation of the tissue cells. Table 2 displays the nature of the observed mutations, and it can be seen that some are functional. Class II HLA typing was different for each subject, but all three patients presented a long lasting (8–15 years) erosive disease. None of the OA patients had detectable mutations in this exon. Due to primer characteristics, antisense sequencing was used as a screening, and a repeated RT-PCR cycle from original RNA and 5′-primer sequencing was used only as a control for mutation presence. In some sequencing experiments, either a faint point mutation was seen in all samples (template-independent systematic error), including the control cDNAs, or all four bases appeared in one position, probably due to inefficient termination reactions, or compression bands appeared in the mutation zone. These experiments were systematically discarded.
Fig. 1.
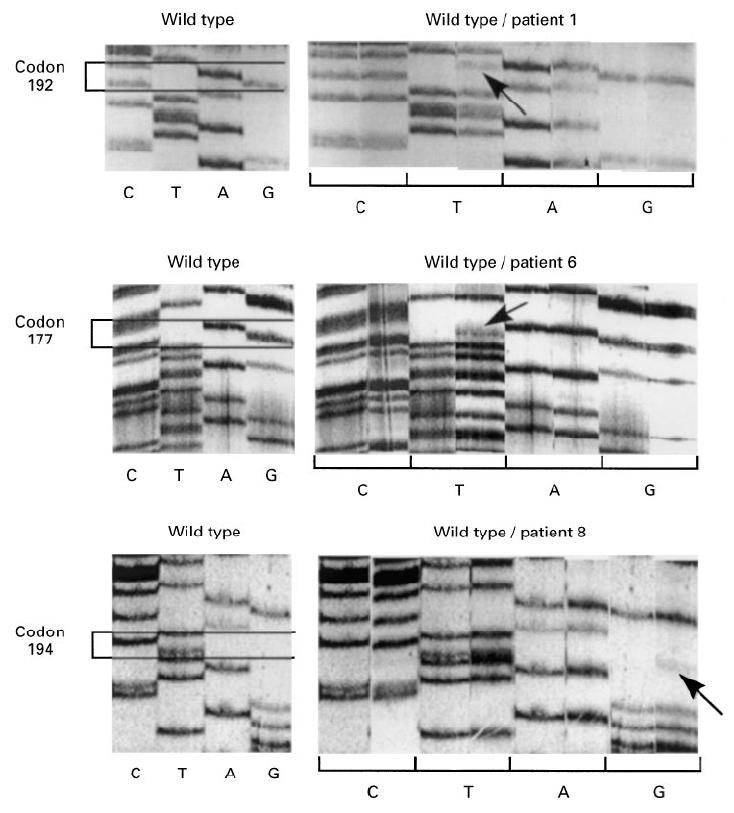
Mutations of p53 exon 6 in total synovial tissue were analysed by di-deoxyDNA-sequencing. For three different RA patients, wild-type codons are presented in the left panels and comparison between wild-type and patient type is depicted in the right panels by base lanes. Coding sense should be read from top to bottom using indicated bases.
Table 2.
p53 exon 6 mutations in RA synovium

Further purification of cell population by adherence to plastic was conducted for an additional four RA tissues, leading mostly to fibroblast-like cells. Exon 6 sequencing was performed on cDNAs prepared from these cell populations, and Fig. 2 clearly shows a point mutation in codon 196 for patient 21. Here again, the band intensity suggests that the mutation is not expressed by a majority of synovial cells, but leads to a stop codon, as underlined in Table 2. In this case, such a mutation could induce p53 accumulation, which was further analysed in immunohistology.
Fig. 2.
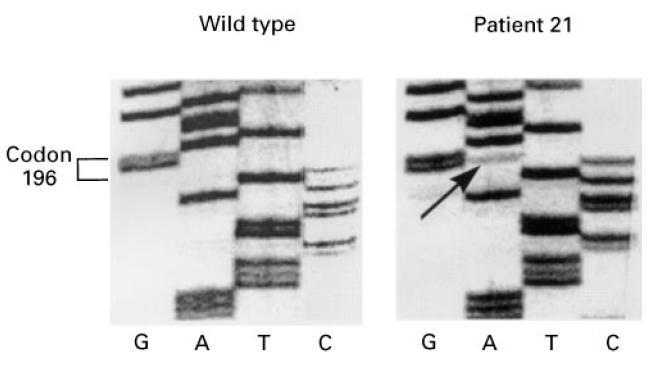
Mutations of p53 exon 6 were further studied in adherent synoviocytes by comparing RA patient 21 sequence with wild-type sequence. Coding sense should be read from top to bottom using indicated bases.
Immunohistological analysis of the p53 protein
In immunohistological studies in normal cells, p53 levels are extremely low owing to the very short half-life of the protein (around 20 min). The mutated protein becomes more stable, prolonging its half-life, so it can be detected by immunoenzymatic techniques. In our hands, the 1801 MoAb was the more efficient to observe a consistent nuclear and sometimes cytoplasmic staining. It stained four out of the 13 tissue biopsies analysed, including patient 21. The distribution of the p53-expressing cells is mostly located in the peripheral lining layer of the tissue, as shown in Fig. 3.
Fig. 3.
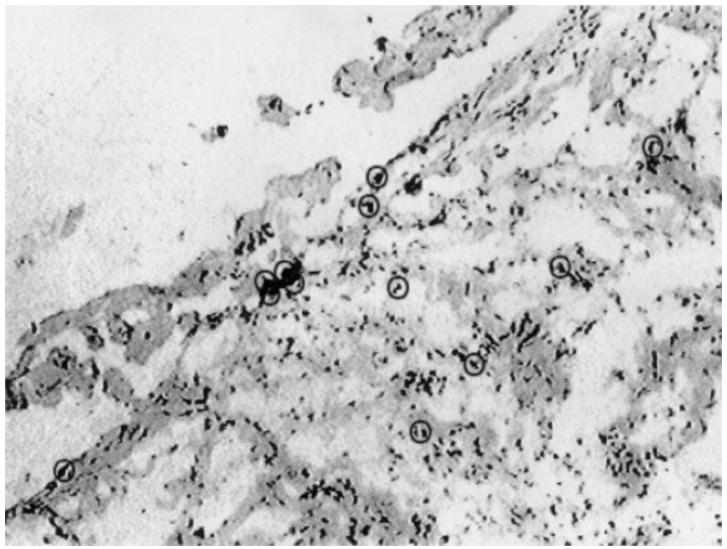
To assess p53 expression in RA synovial tissue by histocytochemistry, a frozen section of RA patient 21 synovial tissue was stained with the 1801 MoAb according to the alkaline phosphatase standard protocol. Circles indicate p53-expressing, red-stained cells, mainly located in the lining layer of the section.
DISCUSSION
Alterations of the p53 tumour suppressor gene have been reported in more than 50% of human cancers [4]. Although p53 mutations have never been detected in normal subjects, they have recently been studied in benign conditions such as psoriasis [10, 11], and so-called chromosome instability syndromes, including ataxia telangiectasia, xeroderma pigmentosum and Bloom's disease [8]. Based on the observation that synovial hyperplasia in RA is associated with proliferation of transformed-appearing synovial lining cells and over-expression of several oncogenes [1], we attempted to determine the role of the p53 tumour suppressor gene in this neoplasia-like situation. A complete study of the p53 tumour suppressor gene was carried out in 24 RA DNAs, looking for gene mutations and protein expression abnormalities. Screened exon 2 and exons 5–9 correspond to the region containing the mutational hot-spot sites described in human cancers [4]. These regions also include the coding sequence for the most conserved amino acid domains of the protein in all species [19]. In this study, mutations of the p53 gene were detected by a combination of PCR, SSCP and sequencing techniques. The method used is sensitive, since diluted mutated DNAs, as low as 10%, can still be easily detected [12]. However, negative SSCP results do not exclude under-represented mutations. It is therefore not excluded that systematic sequencing of the other exons, particularly those containing ‘hot spots’ or frequently mutated regions in human malignancies, could possibly reveal additional mutations.
Preliminary experiments in SSCP of various exons of the p53 protein did not allow exclusion of mutations for exon 6 because of the appearance of faint bands and/or shift. Subsequent dideoxy-sequencing of PCR-amplified DNA from whole synovial tissue for this exon in a population of 20 patients evidenced point mutations for three (15%) patients with erosive, class 3 RA, but none of the three OA patients. However, the proportion of mutated to wild-type bands suggested that a minor cell population was concerned. Because of the transformed-like appearance of synovial fibroblasts, we purified adherent cells from synovial tissue and sequenced the extracted DNA for four additional RA patients. We found a stop codon mutation for a patient with a highly erosive disease. Repeated RT-PCR and sequencing cycles on mutated RNA excluded artefacts induced either by amplification or sequencing. The present results show for the first time p53 mutations in an autoimmune situation, making it possible that abnormalities of the p53 tumour suppressor gene in the fibroblastic/epithelial compartment may play a role in oncogene activation [20] and the characteristic proliferative and destructive features of the rheumatoid synovium in patients with erosive RA. The sample size of the OA patients does not allow exclusion of the possibility that p53 mutations are restricted to inflammatory rheumatic diseases, but underlines the aggressiveness of RA synovial pannus. All the patients presenting mutations had a long-lasting disease (about 8 years duration), and three out of four were treated with methotrexate for a long duration therapy, which could not exclude possible effects of the drug. RA synovial hyperplasia is not a tumour, since p53 is not an oncogene, and synovial sarcoma is extremely rare. In addition, none of our patients expressing a p53 gene mutation presented signs of malignancy after a > 2-year observation period.
We also used an immunohistochemical technique to analyse p53 gene expression. With this procedure, positive staining could indicate the presence of mutations in the p53 gene, since most p53-mutated proteins have a prolonged half-life [7]. Immunohistochemical analysis of the synovium evidenced positive cells for p53 expression, particularly in the lining tissue layer. Such an over-expression in RA synovial tissue has been very recently described [21], suggesting that the apoptosis mechanism could be perturbed in RA synovium, as it is in lupus-like lpr mice [22, 23], and could be more generally in all autoimmune diseases [24], although rheumatoid synoviocytes seem to undergo normal Fas-mediated apoptosis [25]. The presence of p53 staining was in accordance with the presence of p53 gene mutations for patient 21, but not for the three others, where no exon 6 mutations were detectable. However, in these cases p53 protein expression could be due to mutations in other coding regions. In addition, discrepancies between the presence of p53 mutations and the level of protein expression have recently been reported for haematologic cancers [26], suggesting other mechanisms to stabilize the p53 protein, and that immunochemistry cannot predict mutations.
Finally, parallel to a significantly increased tissue expression of the p53 protein, the relatively low frequency of the point mutations observed compared with human malignancies, and their frequently non-functional nature, make unlikely a fundamental role of p53 abnormalities in the aetiology or perpetuation of RA. In addition to p53, deregulation of the cell cycle process and oncogene activation in this disease might derive from other tumour suppressor genes or oncogenes that are currently under investigation.
Acknowledgments
This work was done with grants from INSERM, and the Association de Recherche sur la Polyarthrite, Paris, France. A.T. was recipient of a fellowship from the Consejo Nacional de Investigaciones Científicas y Tecnológicas, Venezuela. We are grateful to Dr Joëlle Simony-La Fontaine and Michelle Radal of the Centre Anticancéreux Val d'Aurelle (Montpellier, France) for expert help in the immunohistological study, to Dr François Canovas for tissue samples, to Jean-Pierre Molès for his advice in optimizing the SSCP technique, and to Michel Jourdan for the XG5 DNA.
References
- 1.Trabandt A, Gay RE, Gay S. Oncogene activation in rheumatoid synovium. APMIS. 1992;100:861–75. doi: 10.1111/j.1699-0463.1992.tb04012.x. [DOI] [PubMed] [Google Scholar]
- 2.Bishop JM. Molecular themes in oncogenesis. Cell. 1991;64:235–48. doi: 10.1016/0092-8674(91)90636-d. [DOI] [PubMed] [Google Scholar]
- 3.Marshall CJ. Tumor suppressor genes. Cell. 1991;64:313–26. doi: 10.1016/0092-8674(91)90641-b. [DOI] [PubMed] [Google Scholar]
- 4.Harris CC, Hollstein M. Clinical implications of the p53 tumor-suppressor gene. New England J Med. 1993;329:1318–27. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- 5.Clarke AR, Purdie CA, Harrison DJ, et al. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–52. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 6.Dittmer D, Pati S, Zambetti G, et al. Gain of function mutations in p53. Nature Genet. 1993;4:42–46. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 7.Oren M, Maltzman W, Levine AJ. Posttranslational regulation of the 54K cellular tumor antigen in normal and transformed cells. Mol Cell Biol. 1981;1:101–10. doi: 10.1128/mcb.1.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu X, Lane DP. Differential induction of transcriptionally active p53 following UV or ionizing radiation: defects in chromosome instability syndromes? Cell. 1993;75:765–78. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- 9.Jonason AS, Kunala S, Price GJ, et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci USA. 1996;93:14025–9. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tadini G, Cerri A, Crosti L, et al. p53 and oncogene expression in psoriasis. Acta Derm Venereol. 1989;146:33–35. [PubMed] [Google Scholar]
- 11.Molès JP, Theillet C, Basset-Seguin N, et al. Mutation of the tumor suppressor gene TP53 is not detected in psoriatic skin. Invest Dermatol. 1993;101:100–2. doi: 10.1111/1523-1747.ep12360920. [DOI] [PubMed] [Google Scholar]
- 12.Kovacs B, Patel A, Hershey JN, et al. Antibodies against p53 in sera from patients with systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum. 1997;40:980–2. doi: 10.1002/art.1780400531. [DOI] [PubMed] [Google Scholar]
- 13.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 14.Togushida J, Yamagushi T, Ritchie B, et al. Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res. 1992;52:6194–9. [PubMed] [Google Scholar]
- 15.Orita M, Iwahana H, Kanazawa H, et al. Detection of polymorphisms of human DNA by gel electrophoresis as single strand conformations polymorphisms. Proc Natl Acad Sci USA. 1989;86:2766–70. doi: 10.1073/pnas.86.8.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordell JF, Falini B, Erber WN, et al. Immunoenzymatic labeling of monoclonal antibodies using immune complexes of alkaline phosphatase and monoclonal anti-alkaline phosphatase (APAAP complexes) J Histochem Cytochem. 1984;32:219–29. doi: 10.1177/32.2.6198355. [DOI] [PubMed] [Google Scholar]
- 17.Mazars GR, Portier M, Zhang XG, et al. Mutations of the p53 gene in human myeloma cell lines. Oncogene. 1992;7:1015–8. [PubMed] [Google Scholar]
- 18.Gaidano G, Ballerini P, Gong JZ, et al. P53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 1991;88:5413–7. doi: 10.1073/pnas.88.12.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soussi T, Caron de Fromentel C, May P. Structural aspects of the p53 protein in relation to gene evolution. Oncogene. 1990;5:945–52. [PubMed] [Google Scholar]
- 20.Dooley S, Helitzka I, Hanselmann R, et al. Constitutive expression of c-fos and c-jun, overexpression of ets-2, and reduced expression of metastasis suppressor gene nm23-H1 in rheumatoid arthritis. Ann Rheum Dis. 1996;55:298–304. doi: 10.1136/ard.55.5.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Firestein GS, Nguyen K, Aupperle KR, et al. Apoptosis in rheumatoid arthritis: p53 overexpression in rheumatoid arthritis synovium. Am J Pathol. 1996;149:2143–51. [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe-Fukunaga R, Brannan CI, Copeland NG, et al. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–7. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 23.Chu JL, Drappa J, Parnassa A, et al. The defect of Fas mRNA expresion in MRL-lpr mice is associated with insertion of the retrotransposon, ETn. J Exp Med. 1993;178:723–30. doi: 10.1084/jem.178.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mountz JD, Wu J, Cheng J, et al. Autoimmune diseases. A problem of defective apoptosis. Arthritis Rheum. 1994;37:1415–20. doi: 10.1002/art.1780371002. [DOI] [PubMed] [Google Scholar]
- 25.Nakajima T, Aono H, Hasunuma T, et al. Apoptosis and functional Fas antigen in rheumatoid arthritis synoviocytes. Arthritis Rheum. 1995;38:485–91. doi: 10.1002/art.1780380405. [DOI] [PubMed] [Google Scholar]
- 26.Martinez-Delgado B, Robledo M, Arranz E, et al. Correlation between mutations in p53 gene and protein expression in human lymphomas. Am J Hematol. 1997;55:1–8. doi: 10.1002/(sici)1096-8652(199705)55:1<1::aid-ajh1>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]