

Abstract
Complement receptor type one (CR1; CD35) binds and processes C3b and C4b opsonized immune complexes and regulates complement activation. We have characterized the epitopes of 13 previously reported and seven new MoAbs to human CR1. The MoAbs formed seven groups based on their reactivity with a panel of deletion forms of CR1. Seventeen of the MoAbs reacted with CR1 at more than one site, a consequence of its repetitive sequence. All five of the MoAbs recognizing epitopes in the nearly identical repeats 3, 10, and 17, as well as one MoAb which reacted with repeats 8 or 1/2 of 9 and 15 or 1/2 of 16, blocked cofactor activity for C3b. Knowledge of the repeats bearing the epitopes for these MoAbs should facilitate the further characterization of CR1.
Keywords: immune adherence, CD35, complement control protein repeats
INTRODUCTION
CR1 is a single-chain type one membrane glycoprotein expressed on most peripheral blood cells, follicular-dendritic cells, Schwann cells and glomerular podocytes [1,2]. CR1, also known as the immune adherence receptor, binds C3b and C4b opsonized immune complexes to erythrocytes and transports them to the spleen and liver for degradation by resident phagocytes [3]. CR1 regulates the activation of the complement cascade by serving as a cofactor for the factor I-mediated cleavage of C3b and C4b [4] and by accelerating the decay of the C3 and C5 convertases [5]. Sequencing of the DNA for the most common allelic form (220 kD, type A or F) revealed that the 1930 amino acid (aa) extracellular domain is composed entirely of 30 repeating subunits, termed short consensus repeats (SCR) or complement control protein repeats (CCP) [6–8]. The carboxy terminus consists of a 25 aa transmembrane region followed by a 43 aa cytoplasmic tail. The repeats are composed of 59–76 aa with four cysteines and one tryptophan conserved. In addition, every seventh repeat is homologous to one another and has the same number of aa and alignment of cysteines and the tryptophan. Homologous repeats share 45–100% aa identity compared with the 20–30% identity of non-homologous repeats. This repetitive homology permits the grouping of every seven contiguous repeats into four larger units, so-called long homologous repeats (LHR A–D) [6]. Every seventh module within CCP repeats 3–18 and 19–28 is 99% and > 90% homologous, respectively, i.e. CCP repeat 3 is homologous to 10 and 17, CCP repeat 4 is homologous to 11 and 18, etc. [6,7,9].
Employing deletion mutants of CR1, herein we locate to CCP repeats the epitopes of seven new and 13 previously reported MoAbs to CR1.
MATERIALS AND METHODS
MoAbs to CR1
MoAbs E11 [10], 7G9 [11], HB8592 [12], 9H3 and 1B4 [13] were provided by R. Taylor (University of Virginia School of Medicine, Charlottesville, VA); 3D9 [13] by E. Brown (Washington University School of Medicine, St Louis, MO); J3B11 and J8B10 [14] by J. Moulds (University of Texas Medical School at Houston, Houston, TX); KuN241 [15] by T. Mohanakumar (Washington University School of Medicine); YZ-1 [16] by R. Jack (Harvard Medical School, Boston, MA); J3D3 [14] was from Amac Inc. (Westbrook, ME); To5 [17] and Ber-MAC-DRC [18] from Dako (Carpenteria, CA). MoAbs 8C9.1, 4D6.1, 4D12.1, 9 A3.1, 3C6.D11, 6B1.H12 and 1F11.G12 were prepared at T Cell Sciences Inc. (Needham, MA). The mouse subclass of the MoAbs was IgG1, except for 8C9.1 which was IgG2b.
Preparation of deletion mutants of CR1
LHR A was obtained by oligonucleotide-directed mutagenesis using Muta-Gene M13 in vitro Mutagenesis Kit (BioRad, Hercules, CA). A translation STOP codon was placed at position 449 (aa numbering of mature protein) of CR1–4 [7], composed of eight and one half CCP. The construct CCP 1–3 was made by replacing aa 195 in LHR A with a STOP codon. ΔCCP 1 (aa 1–60 deleted) and ΔCCP 2 (aa 61–122 deleted) described earlier [19], as well as ΔCCP 3 (aa 123–194 deleted) were made by oligonucleotide-directed mutagenesis. LHR B was obtained from LHR A by site-directed mutagenesis as a result of changing aa in CCP 1–3 into those present in CCP 8–10 [20]. It is composed of aa 451–898. The construct CCP 8–10 was made by replacing aa 645 in the protein LHR B with a STOP codon. The proteins CCP 1–4, composed of aa 1–254, and CCP 8–11, composed of aa 451–704, are by-products of the construction of CCP 1–4 + 15–18 and CCP 8–11 + 15–18, respectively (see below).
To make constructs LHR C and LHR D+, the signal peptide and CCP 15–21 (LHR C), containing aa 898–1353, and CCP 22–30 (LHR D+) containing aa 1349–1928, were amplified by polymerase chain reaction (PCR) using CR1 cDNA in AprM8 (obtained from L. Klickstein, Harvard Medical School) as a template. Although LHR D is composed of CCP 22–28 only, CCP 29 and 30, which are not a part of an LHR, were included in a construct referred to as LHR D+. The signal peptide was then ligated to LHR C and to LHR D+ via restriction sites Avr II and Bam HI, respectively, which were generated by silent mutations. The ligation products were cloned into expression vector pSG5 (Stratagene, La Jolla, CA).
CCP 1–4 + 15–18 and CCP 8–11 + 15–18 were prepared by PCR amplification. CCP 1–4, CCP 8–11 and CCP 15–18 were amplified using CR1–4, CR1–4 (8–9) (obtained from CR1–4 by changing aa of CCP 1–3 to CCP 8–10 [20]) and LHR C, respectively, as templates. CCP 15–18 was then ligated to CCP 1–4 and to CCP 8–11 through an Avr II site, generated by silent mutations, and the constructs cloned into pSG5. CCP 1–4 + 15–18 is composed of aa 1–450 followed by 897–1154. CCP 8–11 + 15–18 contains aa 451–704 followed by 897–1154.
Cell surface and soluble CR1 deletion mutants for flow cytometry and Western blot analyses
Chinese hamster ovary (CHO) cells expressing cell surface CR1 mutants were generated as previously described [21], and included the following cell lines: CCP 1–2 + D, CCP 1–4 + D, CCP 15–16 + D, CCP 15–18 + D, D, and ABCD. Each of the cell surface CR1 mutants included the cytoplasmic, transmembrane, CCP (29–30) and LHR-D regions as well as any additional CCP or LHR domains as indicated. The soluble forms of CR1 used for Western blotting were sCR1 [21,22], sCR1desLHR-A [23], and sCR1desLHR-D. These soluble forms of CR1 lacked the cytoplasmic and transmembrane domains, but included LHR domains ABCD, ACD, BCD, and ABC, respectively, and also included CCP (29–30) in all but the last form, ABC. To establish the location of the 8C9.1 epitope, COS cell supernatants of LHR A, LHR B, ΔCCP 1, ΔCCP 2, ΔCCP 3 were also analysed by Western blotting.
Dot blotting
A BioRad dot blot apparatus and 0.45-μm nitrocellulose membranes were used for most experiments. For some experiments PVDF membranes (Millipore, Bedford, MA) were used. Nitrocellulose membranes were wetted with PBS (1 mm KH2PO4, 10 mm Na2HPO4, 137 mm NaCl, 2.7 mm KCl, pH 7.4). PVDF membranes were prewetted by brief immersion in methanol and rinsed with dH2O followed by PBS. sCR1 (6.25 ng) or deletion mutants of CR1 (10 ng) were applied in a total volume of 200 μl per well. For some experiments sCR1 was reduced by heating in a boiling water bath for 3 min in the presence of 10% 2-mercaptoethanol (2-ME; v/v). Samples were diluted 1:100 with PBS before application. Wells were blocked by gravity filtration of 250 μl 1% bovine serum albumin (BSA; w/v) (Sigma Chemical Co., St Louis, MO) prepared in PBS–0.1% Tween 20 (PBS–T). The blocking solution was filtered through a 2-μm nylon syringe filter before use. For some experiments 1% human serum albumin, chicken egg albumin (Calbiochem, San Diego, CA), or Superblock-PBS (Pierce, Rockford, IL) were used as a blocking protein. Following blocking, wells were washed three times with 300 μl PBS–T under vacuum. Serial four-fold dilutions of the MoAb to anti-CR1, added in a volume of 100 μl/well, were titered versus sCR1 or its deletion mutants (see below) to determine their optimum dilution for assay. Values ranged from 2 ng to 128 ng per well for the monoclonals received as purified immunoglobulin. Ascites samples (HB8592, J3B11, and J8B10) were tested at a dilution of 1:50 000. As a control for non-specific binding, wells without sCR1, but blocked with BSA, were tested. Following gravity filtration and washing, 250 μl of a 1:100 000 dilution (PBS) of horseradish peroxidase (HRP)-conjugated sheep anti-mouse immunoglobulin (Amersham, Arlington Heights, IL) were gravity-filtered through all wells. Wells were washed, as described above, and bound second antibody was visualized by chemiluminescence following the manufacturer's recommendations (Pierce) using Biomax-MR x-ray film (Eastman Kodak, Rochester, NY). Fifteen- and 60-s exposures were made for all blots.
ELISA
ELISA was performed using 3D9 as capture MoAb and E11 conjugated to HRP for detection [19]. For the proteins not recognized by E11, a rabbit polyclonal antibody [21] followed by donkey anti-rabbit IgG–HRP was used. For the proteins not recognized by 3D9, polyclonal anti-CR1 was used as the capture antibody and E11 conjugated to HRP was used for detection. For mapping the 8C9.1 epitope, the antibody was biotinylated with NHS-LC-Biotin (Pierce) and bound antibody was detected with extravidin–HRP (Sigma).
Flow cytometry
The buffer used for flow cytometry was 0.2% BSA, 0.02% NaN3 in PBS. Each of the antibodies to be examined (100 μl at 4 μg/ml in 5% goat serum) was added to CHO cells (100 μl at 2 × 106 cells/ml) expressing various cell surface CR1 mutants and incubated for 30 min on ice. The volume was brought to 3 ml, centrifuged, and the cells were resuspended in 100 μl of diluted FITC-labelled goat anti-mouse immunoglobulin antibody (Southern Biotechnology Associates, Birmingham, AL) in 5% goat serum. After 30 min on ice, the cells were again diluted to 3 ml, centrifuged, and resuspended in 1 ml for analysis on a Becton Dickinson (San Jose, CA) FACScan flow cytometer. Negative controls included the use of an irrelevant isotype-matched MoAb and also a CHO line expressing an irrelevant protein.
Western blot analysis
Each of the soluble forms of CR1 (1 μg) was subjected to non-reducing SDS–PAGE on 4–20% or 10–20% gradient gels followed by transfer to polyvinylidene fluoride membranes (Millipore) using a semidry system. The membranes were blocked for 2 h with 5% dry milk in PBS or TBS (20 mm Tris, 0.5 m NaCl, pH 7.8). The antibodies to be tested (25 μg/ml in 5% goat serum) were incubated with the blocked membranes for 1.5 h with shaking, followed by washing with PBS. Diluted HRP-conjugated goat anti-mouse IgG antibody (Southern Biotechnology) was incubated for 1 h followed by colour development using 4-chloro-1-naphthol (BioRad). For 8C9.1, biotinylated antibody was incubated with the blocked membranes for 1 h at 37°C followed by washing with PBS–T. Detection was by incubation with ExtrAvidin-HRP (Sigma) followed by ECL (Pierce).
Inhibition of cofactor activity
sCR1 (15 ng) was incubated with 750 ng of purified MoAbs or 5 μl of a 1:500 dilution of the ascites samples (HB8592, J3B11, and J8B10) for 30 min at 4°C in a total volume of 10 μl. The samples were transferred to tubes containing 500 ng 125I-C3b (2 × 106 dpm/μg), prepared as previously described [24,25], with or without 100 ng factor I (Quidel, San Diego, CA), and the total volume adjusted to 50 μl with 38–49 μl of 10 mg/100 ml BSA prepared in PBS diluted 1:3 with dH2O. Samples were incubated at 37°C for 45 min and the reaction stopped by the addition of 100 μl of reducing SDS–PAGE loading buffer and heating in a boiling water bath for 5 min. Samples (20 μl) were electrophoresed on a 10% polyacrylamide gel and 125I-labelled C3b cleavage products visualized by autoradiography.
RESULTS
Dot blot analysis
The 20 MoAbs were analysed for binding to a panel of deletion mutants of CR1 (shown in Fig. 1). The differences in aa sequence between homologous CCP repeats of the four LHR are quantified in Fig. 2. The MoAbs formed seven groups based on their reactivity by dot blotting (Table 1). Representative dot blotting results are presented in Fig. 3.
Fig. 1.
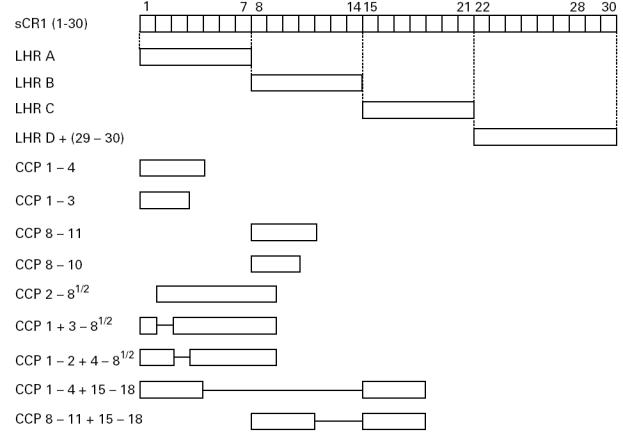
Block diagram of truncated forms of CR1 used for dot blot assays. Soluble CR1 (sCR1) is shown at the top of the figure (complement control protein (CCP) repeats 1–30). Vertical dotted lines identify the ends of long homologous repeats (LHR) A, B, and C; LHR D, CCP repeats 21–28, is linked to repeats 29–30 (denoted as D+ in text). Deletion constructs are shown below their corresponding positions in sCR1. The solid horizontal lines represent deleted repeats.
Fig. 2.
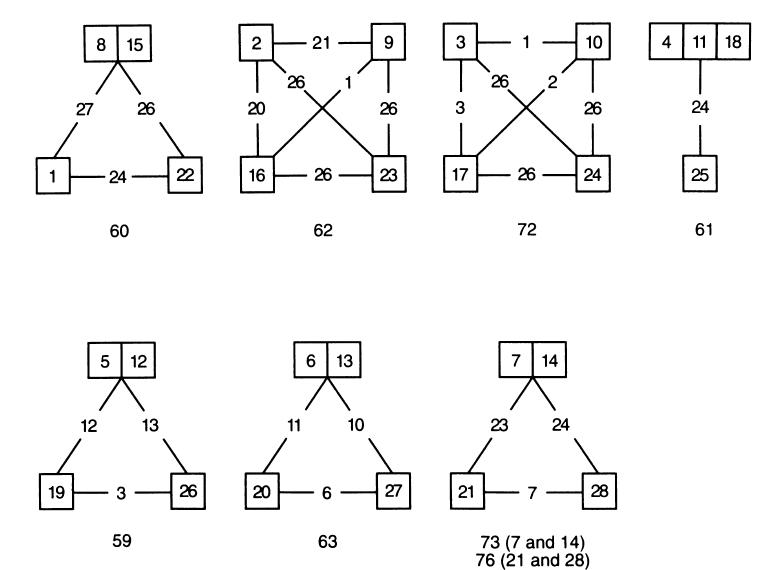
Differences in amino acid sequences between homologous complement control protein (CCP) repeats of long homologous repeats (LHR) A–D. CCP repeats are shown in numbered boxes, each group of four homologous repeats is connected by lines interrupted by the number of aa differences. Abutting repeats have identical aa sequences. The number of aa in the homologous repeats is shown below each group.
Table 1.
Reactivity of MoAbs with sCR1 and its truncated forms by dot blotting

Fig. 3.
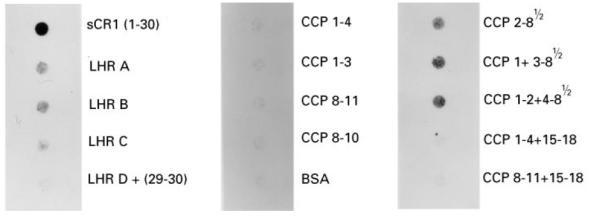
Reactivity of MoAb To5 with sCR1 and its derivatives by dot blotting. To5, 128 ng/well, was assayed for reactivity with sCR1, and the truncated forms. Detection was by chemiluminescence following 15 s and 60 s exposure.
The five MoAbs comprising group 1 reacted with LHR A, B, and C, but not D. Their reactivity with CCP repeats 1–3 and 8–10 further localized the epitopes. Deletion of CCP repeat 3 (but not 1 or 2) from CCP 1–8½ abrogated binding of these MoAbs. Taken together, these results establish repeats 3, 10, and 17 as bearing the epitopes. Figure 2 points out that CCP repeats 3, 10, and 17 differ by not more than three aa.
By similar reasoning, the epitopes for the seven MoAbs in group 2 were expressed in CCP repeats 5–7, 12–14, and 19–21, but not in 26–28. Among these three groups of homologous repeats (i.e. 5, 12, and 19; 6, 13, and 20; and 7, 14, and 21), the group comprising repeats 7, 14, and 21 was less likely to contain the epitopes for group 2 MoAbs due to the 23 aa differences between repeats 7 or 14 versus 21 (Fig. 2).
Group 3 MoAbs E11 and HB8592 reacted with LHR C and D. The lack of reactivity with constructs containing CCP repeats 15–18 restricted the epitope(s) for these MoAbs to CCP repeats 19–21 and 26–28. However, from other experiments reactivity with additional LHR is suggested because E11 immunoprecipitates both LHR A and B ([19] and unpublished observations). In an attempt to demonstrate this reactivity of E11 with LHR A and B, the dot blot protocol was modified by: (i) replacing the blocking agent, BSA, with either human serum albumin, chicken egg albumin, or a commercial blocking agent ‘Superblock’; and (ii) running the standard assay on PVDF rather than nitrocellulose membranes. These modifications did not result in immunorecognition of LHR A or B, although in each case reactivities with LHR C and D were retained (not shown).
Epitopes for group 4 MoAbs KuN241 and 1F11.G12 are in repeats 5–7, and 12–14. Group 5 (J3B11) epitope is within CCP 8 or in the first one-half of repeat 9, as well as in the homologous position in LHR C. The two MoAbs of group 6, 3C6.D11 and 6B1.H12, reacted only with the construct LHR D+ (repeats 22–30).
Using our dot blot assay, we could detect only a weak reaction of MoAbs 8C9.1 with sCR1 and no reaction with any of the deleted forms. As 8C9.1 was of a different isotype (IgG2b), we wondered if the dot blot results were due to poor binding of the second antibody (sheep anti-mouse IgG–HRP) to 8C9.1. We therefore assessed with deletion mutants of CR1 by ELISA and Western blotting using biotinylated 8C9.1. These data were internally consistent and demonstrated that CCP repeat 1 contained the epitope for MoAb 8C9.1, because LHR A from which CCP 1 was deleted was not detected by ELISA or Western blotting (not shown). Moreover, LHR A, from which CCP 2 was deleted, was recognized. Six additional MoAbs (4D6.1, 4D12.1, 9 A3.1, 1F11.G12, 3C6.D11, 6B1.H12) in groups 1, 4, and 6 were also analysed by flow cytometry and Western blotting (not shown), and the results agreed with those by dot blotting.
None of the MoAbs reacted with sCR1 following its reduction (not shown).
Inhibition of cofactor activity
The 20 MoAbs were assessed for their ability to inhibit cofactor activity for the cleavage of 125I-C3b by factor I. A representative analysis of five MoAbs is shown in Fig. 4. The five MoAbs comprising group 1 as well as the one MoAb in group 5, J3B11, blocked cofactor activity. None of the other MoAbs blocked cofactor activity for C3b.
Fig. 4.
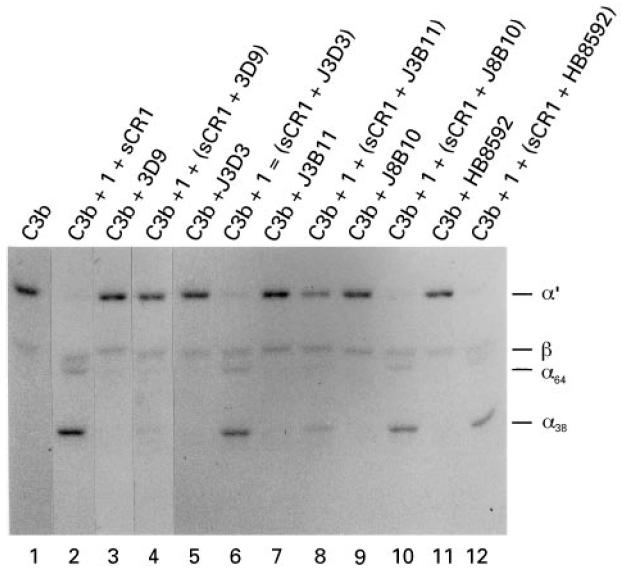
Inhibition of cofactor activity by anti-CR1 MoAbs. J3D3 and 3D9 (750 ng) or 5 μl of a 1:500 dilution of the ascites fluid for J3B11, J8B10, and HB8592 were preincubated with 15 ng of sCR1 and then incubated with 500 ng of 125I-C3b, with or without the addition of 100 ng factor I. Products were separated by SDS–PAGE under reducing conditions and visualized by autoradiography. The antibodies 3D9 (lane 4) and J3B11 (lane 8) completely blocked and partially inhibited, respectively, the cleavage of the α′-chain to 38- and 64-kD fragments. The other three antibodies had no effect (lanes 6, 10 and 12).
DISCUSSION
The purpose of this study was to determine in which LHR and CCP repeats of CR1 epitopes for a panel of 20 MoAbs are located. This was initially assessed by a dot blot procedure using a panel of truncated CR1 constructs. The MoAbs formed seven groups based on their pattern of reactivity (Table 1). Two MoAbs, 8C9.1, which reacted weakly, and E11, which failed to react with two LHR previously noted to possess E11 epitopes ([19] and unpublished observations), were further examined by other methodologies.
Most of the epitopes were localized to LHR A, B and C, although four MoAbs bound to LHR D+ and, of these, two bound only to D+. These epitope locations are consistent with previous reports including: YZ-1 (group 2) binding to LHR A, B, and C, but not D [8]; 1B4 (group 1) binding to the first five CCP repeats of LHR A, B, and C, but not D [26]; and E11 (group 3) recognizing LHR D as well as sites near YZ-1 epitopes [26,27].
The five MoAbs comprising group 1 (epitopes localized to CCP repeats 3, 10 and 17) and the single MoAb defining group 5 (epitope within repeats 8 and 15) were the only MoAbs that inhibited cofactor activity for C3b. These results agree with previous reports identifying 3D9 as blocking this function of CR1 [13]. The ability of J3B11 (group 5) to recognize an epitope in CCP 8 or 9 (and 15 or 16) is consistent with our earlier observations that LHR B and C but not LHR A are the major sites for cofactor activity [20].
Certain MoAbs in group 2 have been reported to block C3b binding (J3D3 and To5), while others (J8B10, 7G9 and YZ-1) do not [14,17]. Also, J3D3 inhibits decay accelerating activity (unpublished observations). Since the epitopes for all of the MoAbs are outside of active sites (CCP repeats 1–4, 8–11, and 15–18), the ability of these MoAbs to block function is surprising. Presumably, by binding near CCP repeats 4, 11 and 18, they cause a steric or conformational effect leading to the inhibition. The lack of inhibition of cofactor activity by J3D3 and To5 may be due to their binding further away from active sites. That J3D3 and To5 inhibit C3b binding only weakly is consistent with this idea [14,28]. A greater concentration of J3D3, compared with J3B11, is required for blockage of C3b-dependent rosetting [14], and To5 is able to bind to CR1 despite the occupancy of its ligand binding sites [28].
Dot blotting failed to identify two of the four E11 binding sites and a binding site for 8C9.1. It may be that the epitope becomes cryptic if the antigen adheres to the membrane. Alternatively, these two MoAbs may have a lower affinity.
The mean number of CR1/cell, especially on erythrocytes, has varied several-fold in the literature [29]. At least part of this problem relates to the multiple binding sites for many MoAbs. Our data provide a simple solution to this problem, since, for example, MoAbs from groups 1–5 can be used as the capture antibody and a MoAb from group 6 employed for detection.
In conclusion, the epitopes for 20 MoAbs to human CR1 have been mapped to CCP repeats. The majority bind to CR1 at multiple (2–4) locations due to CR1's repetitive sequence. None of the MoAbs studied reacted with reduced sCR1. These results should aid in the interpretation and design of quantitative assays for CR1, such as dual antibody ELISA, and in the utilization of these MoAbs for characterization of truncated forms of CR1 in primates [30]. Our results also identify MoAbs which inhibit cofactor activity. Lastly, these data confirm and extend several previous studies of the sites of ligand binding and cofactor activity for C3b.
Acknowledgments
This work was supported in part by funding from the National Institutes of Health (ROI AI37618) and from CytoMed, Inc. (Cambridge, MA). J.P.A. and Washington University have a financial interest in CytoMed, Inc.
References
- 1.Ahearn JM, Fearon DT. Structure and function of the complement receptors, CR1 (CD35) and CR2 (CD21) Adv Immunol. 1989;46:183–219. doi: 10.1016/s0065-2776(08)60654-9. [DOI] [PubMed] [Google Scholar]
- 2.Ross GD. Complement receptor type 1. Curr Top Microbiol Immunol. 1992;178:31–44. doi: 10.1007/978-3-642-77014-2_3. [DOI] [PubMed] [Google Scholar]
- 3.Cornacoff JB, Hebert LA, Smead WL, VanAman ME, Birmingham DJ, Waxman FJ. Primate erythrocyte-immune complex-clearing mechanism. J Clin Invest. 1983;71:236–47. doi: 10.1172/JCI110764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fearon DT. Regulation of the amplification C3 convertase of human complement by an inhibitory protein isolated from human erythrocyte membrane. Proc Natl Acad Sci USA. 1979;76:5867–71. doi: 10.1073/pnas.76.11.5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iida K, Nussenzweig V. Complement receptor is an inhibitor of the complement cascade. J Exp Med. 1981;153:1138–50. doi: 10.1084/jem.153.5.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klickstein LB, Wong WW, Smith JA, Weis JH, Wilson JG, Fearon DT. Human C3b/C4b receptor (CR1): demonstration of long homologous repeating domains that are composed of the short consensus repeats characteristic of C3b/C4b binding proteins. J Exp Med. 1987;165:1095–112. doi: 10.1084/jem.165.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hourcade D, Miesner DR, Atkinson JP, Holers VM. Identification of an alternative polyadenylation site in the human C3b/C4b receptor (CR1) transcriptional unit and prediction of a secreted form of CR1. J Exp Med. 1988;168:1255–70. doi: 10.1084/jem.168.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klickstein LB, Bartow TJ, Miletic V, Rabson LD, Smith JA, Fearon DT. Identification of distinct C3b and C4b recognition sites in the human C3b/C4b receptor (CR1, CD35) by deletion mutagenesis. J Exp Med. 1988;168:1699–717. doi: 10.1084/jem.168.5.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hourcade D, Holers VM, Atkinson JP. The regulators of complement activation (RCA) gene cluster. Adv Immunol. 1989;45:381–416. doi: 10.1016/s0065-2776(08)60697-5. [DOI] [PubMed] [Google Scholar]
- 10.Hogg N, Ross GD, Jones BD, Slusarenko M, Walport MJ, Lachmann P. Identification of an anti-monocyte monoclonal antibody that is specific for membrane complement receptor type 1 (CR1) Eur J Immunol. 1984;14:236–43. doi: 10.1002/eji.1830140307. [DOI] [PubMed] [Google Scholar]
- 11.Reist DJ, Liang HY, Denny D, Martin EN, Scheld WM, Taylor RP. Cross-linked bispecific monoclonal antibody heteropolymers facilitate the clearance of human IgM from the circulation of squirrel monkeys. Eur J Immunol. 1994;24:2018–25. doi: 10.1002/eji.1830240913. [DOI] [PubMed] [Google Scholar]
- 12.Tausk FA, McCluthchen JA, Spechko P, Schreiber RD, Gigli I. Altered erythrocyte C3b receptor expression, immune complexes, and complement activation in homosexual men in varying risk groups for acquired immune deficiency syndrome. J Clin Invest. 1986;78:977–82. doi: 10.1172/JCI112688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Shea JJ, Brown EJ, Seligmann BE, Metcalf JA, Frank MM, Gallin JI. Evidence for distinct intracellular pools of receptors of C3b and C3bi in human neutrophils. J Immunol. 1985;134:2580–7. [PubMed] [Google Scholar]
- 14.Cook J, Fischer E, Boucheiz C, et al. Mouse monoclonal antibodies to the human C3b receptor. Mol Immunol. 1985;22:531–9. doi: 10.1016/0161-5890(85)90176-2. [DOI] [PubMed] [Google Scholar]
- 15.Mathew JM, Naziruddin B, Duffy B, Krych M, Mohanakumar T. Functional analysis of complement receptor 1 using a new monoclonal antibody, KuN241. Hybridoma. 1995;14:29–35. doi: 10.1089/hyb.1995.14.29. [DOI] [PubMed] [Google Scholar]
- 16.Changelian PS, Jack RM, Collins LA, Fearon DT. PMA induces the ligand-independent internalization of CR1 on human neutrophils. J Immunol. 1985;134:1851–8. [PubMed] [Google Scholar]
- 17.Gerdes J, Naiem J, Mason D, Stein H. Human complement (C3b) receptors defined by a mouse monoclonal antibody. Immunol. 1982;45:645–53. [PMC free article] [PubMed] [Google Scholar]
- 18.Yamakawa M, Imai Y. Complement activation in the follicular light zone of human lymphoid tissues. Immunol. 1992;76:378–84. [PMC free article] [PubMed] [Google Scholar]
- 19.Krych M, Hourcade D, Atkinson JP. Sites within the complement C3b/C4b receptor important for the specificity of ligand binding. Proc Natl Acad Sci USA. 1991;88:4353–7. doi: 10.1073/pnas.88.10.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krych M, Clemenza L, Howdeshell D, Hauhart R, Hourcade D, Atkinson JP. Analysis of the functional domains of complement receptor type 1 (C3b/C4b receptor; CD35) by substitution mutagenesis. J Biol Chem. 1994;269:13273–8. [PubMed] [Google Scholar]
- 21.Makrides SC, Scesney SM, Ford PJ, Evans KS, Carson GR, Marsh HC. Cell surface expression of the C3b/C4b receptor (CR1) protects Chinese hamster ovary cells from lysis by human complement. J Biol Chem. 1992;267:24754–61. [PubMed] [Google Scholar]
- 22.Weisman HF, Bartow T, Leppo MK, et al. Soluble human complement receptor type I: in vivo inhibitor of complement suppressing post ischemic myocardial inflammation and necrosis. Sci. 1990;249:146–51. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 23.Scesney SM, Makrides SC, Gosselin ML, et al. A soluble deletion mutant of the human complement receptor type 1, which lacks the C4b binding site, is a selective inhibitor of the alternative complement pathway. Eur J Immunol. 1996;26:1729–35. doi: 10.1002/eji.1830260810. [DOI] [PubMed] [Google Scholar]
- 24.Nickells MW, Ghebrehiwet B, Wetsel RA, Rooney I, Atkinson JP. Complement receptors and membrane regulatory proteins. In: Weir DM, Herzenberg CA, Blackwell C, editors. The handbook of experimental immunology. 5. Cambridge: Blackwell Science, Inc; 1996. pp. 1–16. [Google Scholar]
- 25.Nickells MW, Atkinson JP. Characterization of CR1- and membrane cofactor protein-like proteins of two primates. J Immunol. 1990;144:4262–8. [PubMed] [Google Scholar]
- 26.Bartow TJ, Klickstein LH, Fearon DT. Localization of monoclonal antibody epitopes to CR1 by deletion mutagenesis. Complement Inflamm. 1989;6:312–4. [Google Scholar]
- 27.Bartow T, Klickstein L, Wong W, Roux K, Fearon D. Analysis of the tertiary structure of CR1 by electron microscopy. FASEB J. 1989;3:A501. [Google Scholar]
- 28.Thomsen BS, Nielsen H, Bendixen G. Erythrocyte CR1 determination using monoclonal antibody in a microtiter plate ELISA; receptors are not masked by immune complexes. Allergy. 1986;41:479–86. doi: 10.1111/j.1398-9995.1986.tb00332.x. [DOI] [PubMed] [Google Scholar]
- 29.Quadri RA, Schifferli JA. Over-estimation of the number of complement receptor type 1 (CR1) on erythrocytes. Scand J Immunol. 1992;36:125–30. doi: 10.1111/j.1365-3083.1992.tb02948.x. [DOI] [PubMed] [Google Scholar]
- 30.Nickells MW, Subramanian VB, Clemenza L, Atkinson JP. Identification of complement receptor type 1-related proteins on primate erythrocytes. J Immunol. 1995;154:2829–37. [PubMed] [Google Scholar]