

Abstract
Immunoglobulin gene somatic mutation leads to antibody affinity maturation through the introduction of multiple point mutations in the antigen binding site. No genes have as yet been identified that participate in this process. Bloom's syndrome (BS) is a chromosomal breakage disorder with a mutator phenotype. Most affected individuals exhibit an immunodeficiency of undetermined aetiology. The gene for this disorder, BLM, has recently been identified as a DNA helicase. If this gene were to play a role in immunoglobulin mutation, then people with BS may lack normally mutated antibodies. Since germ-line, non-mutated immunoglobulin genes generally produce low affinity antibodies, impaired helicase activity might be manifested as the immunodeficiency found in BS. Therefore, we asked whether BLM is specifically involved in immunoglobulin hypermutation. Sequences of immunoglobulin variable (V) regions were analysed from small unsorted blood samples obtained from BS individuals and compared with germ-line sequences. BS V regions displayed the normal distribution of mutations, indicating that the defect in BS is not related to the mechanism of somatic mutation. These data strongly argue against BLM being involved in this process. The genetic approach to identifying the genes involved in immunoglobulin mutation will require further studies of DNA repair- and immunodeficient individuals.
Keywords: somatic mutation, immunoglobulin, Bloom's syndrome, variable region, RecQ
INTRODUCTION
Immunoglobulin genes in developing B cells undergo V(D)J gene segment rearrangement to create the primary antibody repertoire composed of highly diverse variable regions that form the antigen binding sites. Upon exposure to antigen and activating T cells, reactive circulating B cells home to germinal centres of peripheral lymphoid organs, where B cell immunoglobulin genes undergo two additional modifications: isotype switching and V region hypermutation [1].
Germ-line encoded, non-mutated antibodies are often of low affinity to the eliciting antigens [1]. Through the process of V region somatic hypermutation, single point mutations occur at high rates throughout the rearranged V region and accumulate over time. Selection for B cells bearing higher affinity antibodies leads to affinity maturation [2,3], so that highly mutated antibodies generally dominate the mature antibody response [2,4]. Mutations tend to accumulate in the hypervariable complementarity-determining regions (CDR) that have been shown to be the antigen contact sites [2,3]. The processes of mutation and selection lead to the clonal expansion of protective antibodies that function efficiently in the immune response. The molecular mechanism of hypermutation remains unclear. Possible mechanisms invoke DNA replication or repair, possibly linked to transcription [5]. None of the proteins involved in the mechanism of hypermutation has as yet been identified.
Bloom's syndrome (BS) is a rare autosomal recessive chromosomal instability disorder in which somatic cells accumulate large numbers of mutations in all loci examined [6]. BS cells exhibit excessive chromosomal breakage and rearrangement with high rates of both sister chromatid exchange and homologous recombination, as well as defects in DNA replication and cell cycle progression. Retardation of the replication fork progress has been reported as well as an abnormal distribution of DNA replicational intermediates [6–10]. Cultured BS cells exhibit an increased sensitivity to DNA-damaging agents that may be attributable to an impairment in DNA replication, repair, or both [11].
The BS gene, BLM, has recently been isolated and a large part of the gene displays significant sequence homologies to the RecQ subfamily of DExH box-containing helicases [11]. The DExH family encompasses a wide variety of proteins and some of them, such as those that are deficient in the diseases xeroderma pigmentosum (XP) and Cockayne's syndrome (CS), are implicated in nucleotide excision and transcription-linked or recombinational DNA repair [12,13]. The helicases are often integral components of larger protein complexes, in which they function as molecular motors in ATP-dependent DNA strand separation [14]. They may also facilitate translocation of the complex along the DNA [14]. The rate of movement at the replication fork is coordinated by the interaction between helicases and the DNA polymerase, where a physical link between enzymes is required to mediate a high rate of progression [14].
One component of the clinical profile in BS is an immune deficiency of unclear aetiology. Affected individuals often develop multiple middle ear and respiratory tract infections beginning in infancy and continuing into childhood. Infections are responsive to antibiotics and are thus presumed to be bacterial. BS B cells have been shown to undergo normal immunoglobulin V(D)J rearrangement [15,16]. Patients have low normal or normal serum levels of IgM, IgG and IgA [17,18]. The chromosomal instability is presumably responsible for the occurrence of a variety of tumours that arise at unusually early ages in a large proportion of BS individuals and renders patients unusually sensitive to chemotherapeutic agents. Other aspects of the syndrome include short stature, male sterility with female subfertility, facial sun sensitivity and hypo- or hyperpigmented skin [6].
We hypothesized that BLM could play a role in immunoglobulin mutation, since the propensity for cancers and for sister chromatid exchanges is probably associated with defects in DNA replication or repair pathways involving this RecQ helicase. If individuals were defective in immunoglobulin hypermutation, the relative paucity of high-affinity antibodies may well be manifested as an immune deficiency in antibody-mediated responses, such as that affecting persons with BS. Here we examine V region sequences in individuals with BS to determine whether their B cells can undergo normal immunoglobulin hypermutation.
The approximately 100 human germ-line VH genes are organized into seven different families based upon sequence homologies. In the present study, we analysed sequences from two well defined heavy chain V region genes, VH6 and VH26. The VH6 family contains only one member with very limited polymorphism [19], simplifying identification of mutations. The VH26 gene, also known as VH18/2, is highly expressed, participates in at least several antimicrobial responses [20] and belongs to the large VH3 gene family. If mutations in these V genes were abnormal, these findings would have implicated the BLM protein in the hypermutation process and it would have helped to define a specific defect in the immunodeficiency found in BS.
SUBJECTS AND METHODS
Study subjects and samples
Blood samples (1–2 ml) were obtained from three individuals with BS (BS 1–3) from the sample bank at the BS Registry at the New York Blood Center. BS 1 is a 22-year-old son of consanguineous parents who has had a history of chronic otitis media requiring myringotomy tube placement. BS 2 is a 12-year-old with a history of multiple infections: recurrent otitis media with myringotomy tube placement, pneumonias, sinusitis and cellulitis. BS 3 is a 21-year-old with recurrent respiratory tract infections. All three subjects had serum levels of IgM and IgG and that were in the low normal range on at least one determination. Blood from each of the BS subjects was stored in liquid nitrogen with an equal volume of cell culture medium with DMSO. The control sample was obtained from a healthy 25-year-old volunteer and was processed immediately.
RNA isolation and reverse transcriptase-polymerase chain reaction
Total cellular RNA was isolated with TRIzol-LS reagent (Gibco BRL, Gaithersburg, MD) according to the manufacturer's specifications. First strand cDNA synthesis was performed with SUPERSCRIPT II (Gibco BRL) as directed by the manufacturer with oligo dT priming using 5–10 μg RNA. Polymerase chain reaction (PCR) was performed with high fidelity Pfu DNA polymerase (Stratagene, La Jolla, CA) and 10% DMSO with 35 cycles at 94°C for 1 min, 56°C for 1 min, 72°C for 2 min. The following PCR primers where used. Two different γ primers were used as the 3′ PCR primer. The 3′ primers were: N-γ aaactcgaggatcgaattcgctctctcggaggtgctcctgga [21] or R-γ aaactcgaggaccttgaccaggcagcccag [22], both of which amplify the first exon of all four γ-chains. The VH6 5′ primer was aaaatcgatctactactactagaggtacagctgcagcagtcaggt [19]; the VH26 5′ primer was aaaatcgatcgccggatcctgtgaggtgcagctgt [21,22]. (Underlined bases were added to facilitate cloning.) Amplified DNA products were gel purified on a 1.2% agarose gel with Qiaex II (Qiagen, Chatsworth, CA) and cloned into the plasmid vector, pCR-Script (Stratagene) at the SrfI site. Transformation was into MutS bacteria (Stratagene) that were mutant for mismatch repair to ensure cloning stability of possible DNA heteroduplex strands.
Hybridization
Bacterial colony hybridization was performed in Rapid-hyb Buffer (Amersham, Cleveland, IL) at 44°C and washed at 46°C. Internal V region probes for VH6 and VH26 with the following sequences were used: for VH26, ctgagactctcctgtgca and for VH6, agaggccttgagtggctg. Hybridizing colonies were selected for sequencing.
Sequence analysis
Plasmid DNA was prepared using Qiagen minipreps. Both DNA strands were sequenced with T3 and T7 primers using an ABI automatic sequencer model 377. Sequences were analysed by Geneworks 2.4 (IntelliGenetics, Mountain View, CA) using germ-line sequences Genbank accession numbers U00509 and J04097 for VH26 and VH6, respectively. The analysis excluded identical sequences obtained from a single subject except to verify the observed mutations. The CDR3 was not analysed due to its hypervariability introduced by VDJ joining. To assign mutations unambiguously and to be certain that the VH genes were correctly identified, we eliminated from the analysis those VH6 or VH26 sequences that contained 20 or more mutations and had three consecutive base changes, similar to the approach taken by Wagner et al. [21]. The remaining sequences with over 20 mutations were excluded if the clone was < 90% identical to the germ-line sequence. The Mann–Whitney non-parametric U-test was used for data analysis.
RESULTS
We examined subjects with BS in whom DNA instability leads to their chromosomal breakage syndrome that includes an immunodeficiency. As none of the proteins involved in immunoglobulin hypermutation are known, we asked whether the BS gene product participates in this process. If so, then the pattern of V region mutations would be abnormal in BS.
V region sequences from three affected individuals were analysed for the presence and nature of mutations and compared with those sequences obtained from a normal volunteer. V region gene sequence analysis using reverse transcriptase (RT)-PCR from RNA from 1–2 ml of stored frozen blood was possible without prior cell sorting of B cell populations, greatly facilitating the procurement of samples from patients with this rare disease. Specific 5′ PCR primers corresponding to different human V gene families allowed for the isolation of two V genes, VH26 or VH6, that were compared with the corresponding germ-line sequences for detection of mutations.
Immunoglobulin gene switching and hypermutation are both T cell-dependent processes that increase with multiple antigenic exposures [2]. The use of γ-hybridizing 3′ PCR primers selectively amplified γ-associated V regions that would be more likely to be mutated; unmutated V genes associated with μ constant regions from naive circulating B cells would be avoided. Colony hybridization of cloned PCR products was performed with V region probes under non-stringent conditions so as not to exclude mutated sequences.
VH26 analysis
Results of RT-PCR amplification with the VH26 primer yielded several sequences, all of which were mutated in both the control and in the BS 1 samples when compared with the germ-line VH26 sequence (Table 1). Representative sequences from BS 1 are shown in Fig. 1. By the rigorous exclusion criteria described above, six of the 19 sequences were not definitively VH26 genes; four from the control and two from BS 1. Four independent sequences from the control subject VH26 sequence analysis were compared with nine from BS 1 in Table 1. The mean number and range of mutations per V region were similar (P = 0.94), with a mean of 13 mutations for the control and 13 for BS 1, with ranges of 9–18 and 1–23, respectively. The distribution of mutation throughout the 292 bases analysed appeared normal, as there were higher proportions of mutations found in the CDRs (Fig. 1). While 23% of total VH26 bases were in the CDRs, these were preferentially mutated in both the control and BS 1 sequences, with 60% of the total mutations in the control and 46% in BS 1. Transition mutations were more frequently seen than transversions in somatically mutated V regions, whereas an unbiased ratio of transitions versus tranversions would be 0.5. Here, the transition/transversion ratios were 1.36 for the control and 1.24 for BS 1, consistent with the normal preponderance of transitions seen in mutated V regions [23]. The ratio of replacement to silent mutations, or R/S ratio, was 1.89 for the control and 2.6 for BS 1. The R/S ratio in the CDRs was 3.57 in the control and 7.83 in BS 1. As is generally found for V region mutations, multiple single base substitutions were far more frequent than deletions or insertions [2]. Subject BS 1 had an in-frame, three base-pair deletion in one VH26 gene.
Table 1.
Analysis of V region mutations in Bloom's syndrome (BS)
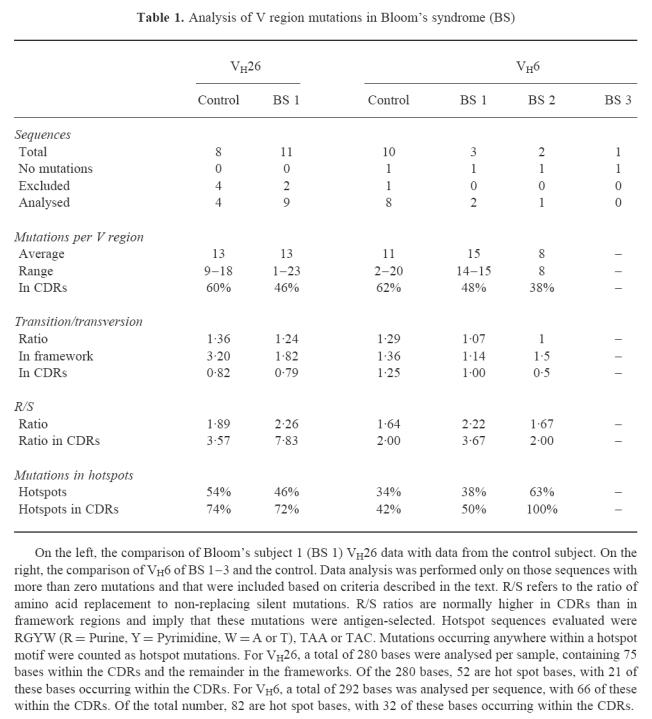
Fig. 1.

Persons with Bloom's syndrome (BS) have normal VH26 mutations. Nucleotide sequences of mutated VH26 regions from peripheral blood lymphocytes of an individual with BS. Sequences are labelled 1a–1f, referring to independent bacterial colonies with cloned polymerase chain reaction (PCR) products derived from BS 1. All clones were associated with IgG rearrangements. Sequences not clearly attributable to VH26, based on criteria described in the text, are not shown and were not analysed. CDR1 and CDR2 are as indicated. Dashes indicate positions with sequence identity to the germ-line. Periods indicate positions of deletion seen in BS sequence 1a only; note that this does not result in a frameshift. The sequences start at the VH26 PCR primer sequence. Genbank accession numbers have been assigned: no. AF015123-35.
The occurrence of mutations within V regions is not random. Analysis of many mutations from a variety of anti-hapten responses has demonstrated that there exist preferred sites of mutation, or ‘hotspots’ [23–25]. One such hotspot is the second base (G) of the serine codon AGC/T [23,24] within the general motif of RGYW (R = Purine, Y = Pyrimidine; W = A or T) or in the sequence TAA or TAC [24,25]. Table 1 shows that a higher proportion of the mutations seen were in hotspots (46–54%) than can be accounted for their total number (28%). Most mutations within the CDRs for VH26 were found in the hotspot sequences (RGYW, TAA or TAC), even though these hotspots represent ≈ 50% of the CDR sequences.
Sequences from subject BS 1 represent normal hypermutation, including the presence of a small genealogy found in bacterial clones d and e (Fig. 1). These two sequences share 12 common mutations, with multiple other unique mutations contained within each clone. Since these two sequences also share the same CDR3 (data not shown), they are likely to have come from B cells derived from a common B cell precursor. These data are similar to those often found in hybridoma genealogies [3,26], probably represent a normal immune response and are probably real mutations and not genetic polymorphisms.
VH6 analysis
Analysis of the VH6 (Table 1 and Fig. 2) sequences shows results similar to those obtained from the VH26 data. Here, sequences were analysed from three BS subjects, BS 1, BS 2 and BS 3. Fewer VH6 sequences were obtained in comparison with VH26, consistent with its lower level of expression in circulating B cells [27]. Only one of the control and none of the BS VH6 sequences were excluded from the analysis. Of the two distinct mutated VH6 sequences obtained from BS 1, there were 14 and 15 mutations, while the one mutated VH6 sequence from BS 2 contained eight mutations. Of the VH6 sequences obtained from the control and from patients, one sequence from each contained no mutations. This includes the only VH6 sequence obtained from BS 3. These sequences are not likely to have come from naive B cells, since they were associated with a switched γ isotype. Perhaps the VH6 V region can function adequately in the germ-line state in some B cells.
Fig. 2.

Persons with Bloom's syndrome (BS) have normal VH6 mutations. Nucleotide sequences of mutated VH6 regions from peripheral blood lymphocytes of persons with BS, BS 1–3. Two independent bacterial colonies from subject BS 1 are labelled as 1a and 1b. As with VH26, all clones were associated with IgG rearrangements, with CDR1 and CDR2 as shown. As with VH26, sequences not attributed to VH6 are not shown and were not analysed. Dashes indicate positions with sequence identity to the germ-line. No VH6 clones were excluded. The sequences start 3′ of the 5′ VH6 polymerase chain reaction (PCR) primer at amino acid 9. Genbank accession numbers for these sequences have been assigned: no. AF015136–46.
The analysis of the eight mutated control sequences revealed an average of 11 mutations, with a range of 2–20 per V region that were similar to BS 1 (P = 0.73) and BS 2 (P = 0.22). All mutations were single base substitutions. The CDRs comprised 27% of the total bases analysed and, as for VH26, contained a disproportionate number of the total mutations: 62%, 48% and 38% for the control, BS 1 and BS 2, respectively. As with VH26, transitions were preferred over transversions; ratios for the control and the two BS patients were 1–1.36. Hotspots were more commonly mutated; they comprised 19% of the total bases, yet represented 34% of the control's and 38% or 63% of the BS subjects' mutated bases. In the single sequence obtained from BS 2, all eight of the mutations were in hotspots within CDR1 and CDR2.
DISCUSSION
This study represents an attempt to identify a protein normally utilized in immunoglobulin hypermutation. The rationale for the study of individuals with BS was to examine the phenomenon in persons known to be genetically immunodeficient in whom cells exhibit DNA instability. Mutations in the V regions from BS subjects 1 and 2 were similar to those found from the control sample, including the number, distribution and nature of mutations. Mutations were characteristic of those found in murine and human immune responses: they were often located in the mutational hotspots, were predominately replacement substitutions in the CDRs, were more commonly transitions and appeared preferentially in hotspot motifs. There were no significant differences in the number of consecutive mutations, or doublets, seen in patients and controls (data not shown). This analysis was restricted to productive immunoglobulin V genes in presumably selected B cells. DNA breakage could theoretically lead to a higher than normal frequency of non-productive rearrangements which would not be detected by our methods. In light of independent reports of normal V(D)J joining [15,16] and near normal serum IgG and IgA in most BS individuals [17,18], however, our data probably accurately reflect V region sequences.
We conclude that immunoglobulin gene hypermutation is normal in BS. Therefore, the BLM gene product is not essential for somatic hypermutation. These results do not exclude the possibility that helicases other than BLM may play a role in mutation. Defining the mechanism of immunoglobulin mutation at the molecular level awaits the identification of at least some of the proteins involved. Possible mechanisms include error-prone DNA repair [28], the introduction of mutations in the lagging strand of replicating immunoglobulin genes that are then incorporated into the genome [29], or activity of a ‘mutator’ protein that could interact with the transcription initiation complex [5] or participate in RT-linked mutation [30]. Any of these models may implicate helicase activity to account for DNA accessibility required for base replacement. The function of these proteins may be within the base excision process itself or in the resolution of altered DNA sequences.
VH26 has been shown to be involved in a variety of immune responses, including against bacterial capsid antigens [20] that may be relevant to the respiratory infections seen in patients with BS. From the data presented here and elsewhere [15–18], immunoglobulin genes in BS undergo normal isotype switching, VDJ recombination and somatic mutation, and these antibodies are secreted in normal or near normal amounts. Hence, susceptibility to bacterial infections in BS patients is probably not attributable to an intrinsic B cell defect.
The RecQ family of genes that includes BLM contains additional members that may be involved in DNA repair and possibly in somatic mutation. The Werner's syndrome gene, WRN, also contains helicase motifs [31] and, like BLM, mutations in this gene cause mutator phenotypes [32]. The yeast homologue of BLM, Sgs1, is a helicase that interacts with topoisomerases I and II and has been implicated in chromosome separation during replication [33–35]. Topoisomerase II was expressed at a low level in one BS patient tested [36], suggesting an additional molecular function for BLM. Topoisomerase II functions in replication, transcription and chromosomal segregation to assist in topologic interconversions via double-stranded DNA breaks. The chromosomal breaks, gaps and translocations and the intra- and interchromosomal strand exchanges found in BS cells point to a disturbance of DNA replication [32]. The genomic instability found in BS and in Werner's syndrome may not be due solely to loss of helicase activity, since WRN and Sgs1 mutations have been found downstream of their helicase domains [31,35]. Evolutionary conservation of these genes is consistent with their playing a role in the preservation of genomic integrity.
Human DNA-repair genes include those that are defective in XP, including the XPD and XPB helicases [21,37], and the CS helicase-like ERCC6/CSB genes [37]. Cells from persons with these genetic defects exhibit defects in nucleotide excision repair [12,13]. Peripheral blood samples [21] or B cell lines [37] obtained from individuals with some XP and CS complementation groups have been found to contain normal B cell V region mutations. Given the inability thus far to find human (or mouse) syndromes associated with a lack of immunoglobulin mutation, perhaps the factors that are responsible for somatic mutation may not be genes that are known to be directly involved in DNA repair. Alternatively, perhaps these factors are so critical as to be indispensable for human development, and thus mutations are lethal. These questions may best be addressed by similar studies of individuals with DNA repair defects such as Werner's syndrome and CS, ataxia-telangiectasia, Wiskott–Aldrich and additional XP complementation groups.
Acknowledgments
The authors would like to thank Matthew D. Scharff for his support and Nathan Ellis and Arturo Casadevall for useful discussions. The Einstein Cancer Center sequencing and oligonucleotide synthesis facilities provided invaluable services. N.S.G. was supported by NIH 5K11CA01635, the James McDonnell Foundation and the American Society of Hematology, S.Z.S. by NIHT32GM07288 and J.G. by NIH CA50897.
References
- 1.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–8. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 2.Berek C, Milstein C. Mutation drift and repertoire shift in the maturation of the immune response. Immunol Rev. 1987;96:23–41. doi: 10.1111/j.1600-065x.1987.tb00507.x. [DOI] [PubMed] [Google Scholar]
- 3.Casson LP, Manser T. Random mutagenesis of two complementarity determining region amino acids yields an unexpectedly high frequency of antibodies with increased affinity for both cognate antigen and autoantigen. J Exp Med. 1995;182:743–50. doi: 10.1084/jem.182.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelsoe G. Life and death in germinal centers (redux) Immunity. 1996;4:107–11. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- 5.Storb U, Peters A, Klotz E, Rogerson B, Hackett J. The mechanism of somatic hypermutation studied with transgenic and transfected target genes. Semin Immunol. 1996;8:131–40. doi: 10.1006/smim.1996.0017. [DOI] [PubMed] [Google Scholar]
- 6.German J. Bloom syndrome—a mendelian prototype of somatic mutational disease. Med. 1993;72:393–406. [PubMed] [Google Scholar]
- 7.Chaganti RS, Schonberg S, German J. A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc Natl Acad Sci USA. 1974;71:4508–12. doi: 10.1073/pnas.71.11.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poot M, Hoehn H, Nicotera TM, Rudiger HW. Cell kinetic evidence suggests elevated oxidative stress in cultured cells of Bloom's syndrome. Free Radic Res Commun. 1989;7:179–87. doi: 10.3109/10715768909087940. [DOI] [PubMed] [Google Scholar]
- 9.Hand R, German J. A retarded rate of DNA chain growth in Bloom's syndrome. Proc Natl Acad Sci USA. 1975;72:758–62. doi: 10.1073/pnas.72.2.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lonn U, Lonn S, Nylen U, Winblad G, German J. An abnormal profile of DNA replication intermediates in Bloom's syndrome. Cancer Res. 1990;50:3141–5. [PubMed] [Google Scholar]
- 11.Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Clocci S, Proytcheva M, German J. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–66. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 12.Sung P, Bailly V, Weber C, Thompson LH, Prakash L, Prakash S. Human xeroderma pigmentosum group D gene encodes a DNA helicase. Nature. 1993;365:852–5. doi: 10.1038/365852a0. [DOI] [PubMed] [Google Scholar]
- 13.Troelstra C, van Gool A, de Wit J, Vermculen W, Bootsma D, Hoeijmakers JH. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell. 1992;71:939–53. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- 14.West SC. DNA helicases: new breeds of translocating motors and molecular pumps. Cell. 1996;86:177–80. doi: 10.1016/s0092-8674(00)80088-4. [DOI] [PubMed] [Google Scholar]
- 15.Petrini JH, Donovan JW, Dimare C, Weaver DT. Normal V(D)J coding junction formation in DNA ligase I deficiency syndromes. J Immunol. 1994;152:176–83. [PubMed] [Google Scholar]
- 16.Hsieh CL, Arlett CF, Lieber MR. V(D)J recombination in ataxia telangiectasia, Bloom's syndrome, and a DNA ligase I-associated immunodeficiency disorder. J Biol Chem. 1992;268:20105–9. [PubMed] [Google Scholar]
- 17.Kondo N, Ozawa T, Kato Y, Motoyoshi F, Kasahara K, Kameyama T, Orii T. Reduced secreted mu mRNA synthesis in selective IgM deficiency of Bloom's syndrome. Clin Exp Immunol. 1992;88:35–40. doi: 10.1111/j.1365-2249.1992.tb03035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondo N, Motoyoshi F, Mori S, Kuwabara N, Orii T, German J. Long-term study of the immunodeficiency of Bloom's syndrome. Acta Paediatr. 1992;81:86–90. doi: 10.1111/j.1651-2227.1992.tb12088.x. [DOI] [PubMed] [Google Scholar]
- 19.Chu YW, Marin E, Fuleihan R, Ramesh N, Rosen FS, Geha RS, Insel RA. Somatic mutation of human immunoglobulin V genes in the X-linked hyperIgM syndrome. J Clin Invest. 1995;95:1389–93. doi: 10.1172/JCI117791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adderson EE, Shackelford PG, Quinn A, Wilson PM, Cunningham MW, Insel RA, Carroll WL. Restricted immunoglobulin VH usage and VDJ combinations in the human response to Haemophilus influenzae type b capsular polysaccharide. Nucleotide sequences of monospecific anti-Haemophilus antibodies and polyspecific antibodies cross-reacting with self antigens. J Clin Invest. 1993;91:2734–43. doi: 10.1172/JCI116514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner SD, Elvin JG, Norris P, McGregor JM, Neuberger MS. Somatic hypermutation of Ig genes in patients with xeroderma pigmentosum (XP-D) Int Immunol. 1996;8:701–5. doi: 10.1093/intimm/8.5.701. [DOI] [PubMed] [Google Scholar]
- 22.Klein U, Kuppers R, Rajewsky K. Variable region gene analysis of B cell subsets derived from a 4-year-old child: somatically mutated memory B cells accumulate in the peripheral blood already at young age. J Exp Med. 1994;180:1383–93. doi: 10.1084/jem.180.4.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neuberger MS, Milstein C. Somatic hypermutation. Curr Opin Immunol. 1995;7:248–54. doi: 10.1016/0952-7915(95)80010-7. [DOI] [PubMed] [Google Scholar]
- 24.Rogozin IB, Kolchanov NA. Somatic hypermutagenesis in immunoglobulin genes. II. Influence of neighbouring base sequences on mutagenesis. Biochim Biophys Acta. 1992;1171:11–18. doi: 10.1016/0167-4781(92)90134-l. [DOI] [PubMed] [Google Scholar]
- 25.Smith DS, Creadon G, Jena PK, Portanova JP, Kotzin BL, Wysocki LJ. Di- and trinucleotide target preferences of somatic mutagenesis in normal and autoreactive B cells. J Immunol. 1996;156:2642–52. [PubMed] [Google Scholar]
- 26.Clarke SH, Staudt LM, Kavaler J, Schwartz D, Gerhard WU, Weigert MG. V region gene usage and somatic mutation in the primary and secondary responses to influenza virus hemagglutinin. J Immunol. 1990;144:2795–801. [PubMed] [Google Scholar]
- 27.Fumoux F, Guigou V, Blaise D, Maraninchi D, Fougereau M, Schiff C. Reconstitution of human immunoglobulin VH repertoire after bone marrow transplantation mimics B-cell ontogeny. Blood. 1993;81:3153–7. [PubMed] [Google Scholar]
- 28.Brenner S, Milstein C. Origin of antibody variation. Nature. 1966;211:242–3. [PubMed] [Google Scholar]
- 29.Manser T. The efficiency of antibody affinity maturation: can the rate of B-cell division be limiting? Immunol Today. 1990;11:305–8. doi: 10.1016/0167-5699(90)90124-r. [DOI] [PubMed] [Google Scholar]
- 30.Steele EJ, Rothenfluh HS, Blanden RV. Mechanism of antigen-driven somatic hypermutation of rearranged immunoglobulin V(D)J genes in the mouse. Immunol Cell Biol. 1997;75:82–95. doi: 10.1038/icb.1997.12. [DOI] [PubMed] [Google Scholar]
- 31.Yu CE, Oshima J, Fu YH, et al. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–62. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 32.Ellis NA. Mutation-causing mutations. Nature. 1996;381:110–1. doi: 10.1038/381110a0. [DOI] [PubMed] [Google Scholar]
- 33.Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R. The yeast type I topoisomerase Top3 interacts with Sgsl, a DNA helicase homolog—a potential eukaryotic reverse gyrase. Mol Cell Biol. 1994;14:8391–8. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watt PM, Louis EJ, Borts RH, Hickson ID. Sgsl: a eukaryotic homolog of E. coli Recq that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell. 1995;81:253–60. doi: 10.1016/0092-8674(95)90335-6. [DOI] [PubMed] [Google Scholar]
- 35.Lu J, Mullen JR, Brill SJ, Kleff S, Romeo AM, Sternglanz R. Human homologues of yeast helicase. Nature. 1996;383:678–9. doi: 10.1038/383678a0. [DOI] [PubMed] [Google Scholar]
- 36.Foucault F, Vaury C, Barakat A, Thibout D, Planchon P, Jaulin C, Praz F, Amor-Gueret M. Characterization of a new BLM mutation associated with a topoisomerase IIα defect in a patient with Bloom's syndrome. Human Molec Genet. 1997;6:1427–34. doi: 10.1093/hmg/6.9.1427. [DOI] [PubMed] [Google Scholar]
- 37.Nayun K, Kage K, Matsuda F, Lefranc MP, Storb U. B lymphocytes of xeroderma pigmentosum or Cockayne syndrome patients with inherited defects in nucleotide excision repair are fully capable of somatic hypermutation of immunoglobulin genes. J Exp Med. 1997;186:413–9. doi: 10.1084/jem.186.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]