

Abstract
Patients with IgA deficiency often demonstrate circulating antibodies against IgA, which have been suggested to be associated with transfusion reactions. Sera from three patients with common variable immunodeficiency (CVID) and one with a selective IgA deficiency with anti-IgA antibodies receiving subcutaneous gammaglobulin replacement therapy were analysed for serum levels of IgG, IgA and anti-IgA before and during a treatment period of 4–7 years. Treatment with gammaglobulin preparations containing significant amounts of IgA ( < 5 mg/ml) resulted in a decrease or disappearance of the anti-IgA antibodies. Analysis of serum fractions, however, revealed anti-IgA activity in the complex-containing fractions. In vitro experiments gave similar results with a shift of anti-IgA activity from the monomeric to the complex-containing fractions (that could not be detected in whole serum). When the patients were subsequently switched to treatment with a preparation containing less IgA ( < 80 μg/ml) or made an interruption in the treatment schedule, the anti-IgA antibodies reappeared. Importantly, however, one of the patients lost his anti-IgA activity during a 3-month period on the preparation containing the higher IgA levels, and these antibodies did not reappear after switching to the low IgA-containing preparation. After 5 years on this preparation, anti-IgA can still not be detected, suggesting induction of unresponsiveness.
Keywords: anti-IgA, IgA deficiency, subcutaneous gammaglobulin, tolerization, immune complexes
INTRODUCTION
Treatment of patients with hypogammaglobulinaemia with immunoglobulin has been used since Bruton first described congenital agammaglobulinaemia [1]. Since then, various strategies for immunoglobulin replacement have been employed.
Among immunodeficient patients, there are a number of individuals with low or undetectable levels of IgA. The prevalence of anti-IgA antibodies among patients (Caucasian) with IgA deficiency is around 30–40%. In patients with combined IgA-IgG2 deficiency the proportion rises to 50–60% [2,3] and the presence and level of anti-IgA in IgA-deficient blood donors is remarkably constant over time [4].
The major treatment for these patients has been infusion with immunoglobulin. A few patients have been reported to experience severe side effects, some of which are probably caused by anti-IgA antibodies [5–8]. Subcutaneous immunoglobulin infusion has been proven to be a safe and less expensive choice, with almost no adverse effects and a possibility of home therapy [9]. It was noted that the titres of anti-IgA were markedly reduced or eliminated in some of our treated patients. In the present study we have tried to find a plausible explanation for the dramatic changes in anti-IgA activities seen during treatment with subcutaneous immunoglobulin infusions.
MATERIALS AND METHODS
Patients
From a total of 194 patients with selective IgA deficiency ( < 50 μg/ml) and 48 patients with hypogammaglobulinaemia, we selected four patients with disappearance of anti-IgA antibodies during treatment with subcutaneous immunoglobulin. All patients had been followed for 4–7 years (see Tables 1 and 2 for details). Three of the patients, two males and one female (patients 1, 2 and 3) had hypogammaglobulinaemia and complete IgA deficiency and one male patient (patient 4) had a selective IgA deficiency.
Table 1.
Patients with immunodeficiencies
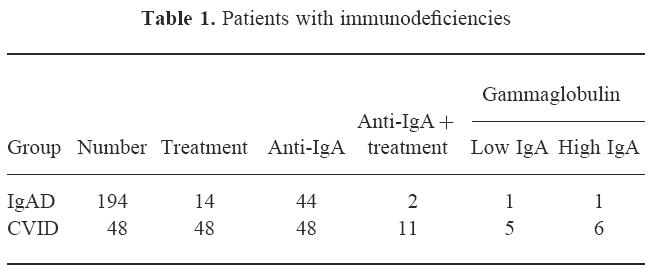
Table 2.
IgG, IgA, and anti-IgA activity in sera from patients with IgA deficiencies before and during subcutaneous immunoglobulin infusion therapy

Blood samples were collected before commencement of the immunoglobulin treatment and at regular intervals thereafter. The dose of immunoglobulin was 100 mg/kg per week. We used two different commercially available preparations with varying contents of IgA: Gammabulin containing < 3% IgA (4.8 mg/ml) (Immuno, Sweden), and Gammaglobulin containing < 0.01% IgA (82 μg/ml) (Pharmacia-Upjohn, Uppsala, Sweden). The latter preparation contained very low levels of IgA that have been shown to be tolerated in patients with high levels of anti-IgA antibodies [9,10].
Where appropriate, control sera from healthy blood donors were included in the study.
Immunoglobulin quantification
Concentrations of IgG, IgA and IgM were measured using rate nephelometry (Beckman Instruments, Brea, CA), ELISA (in-house method) and radial immunodiffusion with commercial immunodiffusion plates (Behringwerke AG, Marburg, Germany). The validity of the measurements was controlled by calibration against a commercial serum preparation traceable to an internationally recognized reference preparation (CRM 470) [11].
Anti-IgA
The activity of anti-IgA antibodies was determined both by a haemagglutination assay based on the agglutination of human erythrocytes (0, Rh-negative) coated with purified IgA, and an ELISA system based on plates coated with purified IgA as described in Hammarström et al. [12]. The results for anti-IgA activity in ELISA, with a serum dilution of 1:100, were semiquantitatively graded. The ranges were as follows: optical density (OD) > 2.0 = strong positive (+ + +), 1.0–2.0 = positive (+ +), 0.4–1.0 = weakly positive (+), < 0.4 = negative (−). The purified IgA was prepared essentially as described by Hiemstra et al. [13] or purchased from Sigma (St Louis, MO). The results obtained using these two preparations were nearly identical. The positive reference serum used was from a patient with IgA deficiency and a high titre of anti-IgA antibodies.
Serum fractionation
Serum components were fractionated according to their molecular size by use of a gel filtration column connected to an automated chromatography system (Superose 6, FPLC; Pharmacia-Upjohn). The column was calibrated by separation and fraction analysis of normal sera and monomeric and heat-aggregated purified IgG. Fractions were collected and analysed for protein, immunoglobulin isotypes and IgG anti-IgA activity. Care was taken in normalizing all column fractions against dilution effects.
In vitro complexing
In vitro complexes were formed by the addition of purified IgA to sera containing anti-IgA antibodies with no detectable IgA. The IgA was added to give the concentrations 1, 0.1 and 0.01 mg IgA/ml. After incubation for 60 min followed by column fractionation, IgG, IgA levels and anti-IgA activity were measured both in the original mixtures and in the fractions.
RESULTS
Persistence of anti-IgA antibodies in IgA-deficient patients
We detected anti-IgA activity in 44 (23%) out of 194 patients with IgA deficiency ( < 50 μg/ml). Of these 44 patients, 40 could be followed for a total of 560 patient years without treatment. Only four untreated patients turned negative for anti-IgA, and they were all weakly positive in the first sample obtained. None of the patients with moderate/strong positive anti-IgA reactivity converted.
Disappearance of strong anti-IgA reactivity
Two patients with IgA deficiency and anti-IgA antibodies underwent treatment with immunoglobulin, one receiving the low IgA-containing preparation ( < 80 μg/ml), the other receiving the preparation containing up to 5 mg/ml of IgA (Table 1). With the low IgA preparation the anti-IgA activity remained constant over the observation period of 9 years, while with the high IgA preparation the anti-IgA reached undetectable levels within 1 month of starting therapy (patient 4) (Tables 1 and 2). In the CVID group of treated patients, 11 had anti-IgA antibodies (Table 1). Of these, four patients remained stable in their anti-IgA activity and they received the low IgA preparation. Five patients lost their anti-IgA activity during therapy with the high IgA preparation (patients 1, 2 and 3) (Tables 1 and 2). The remaining two patients demonstrated a gradual decrease in anti-IgA activity, one on the low, the other on the high IgA-containing preparation.
The results obtained from analysis of IgG, IgA and anti-IgA in sera from four of the above patients, taken before and at various time points during treatment, are summarized in Table 2.
In the first sample, taken before commencement of treatment, sera from all four patients were strongly positive for anti-IgA and IgA was not detectable using nephelometry, immunodiffusion ( < 20 μg/ml) or ELISA ( < 100 ng/ml). The levels of IgA increased and the anti-IgA decreased after treatment with an immunoglobulin preparation containing up to 5 mg/ml of IgA, while a preparation with up to 80 μg/ml IgA did not induce these effects (Table 2).
Patient 4 (selective IgA deficiency) had an interruption of his treatment during which the IgA level declined and the anti-IgA reappeared. This effect was reversed when treatment was resumed.
When patient 2 was switched to the low IgA ( < 80 μg/ml) preparation, serum IgA was no longer detectable without reappearance of anti-IgA. This situation has remained unchanged for 5 years (Table 2). Similar changes were observed in patient 3, although here the observation period was only 1 year (Table 2).
Fractionation of serum components by gel filtration
A typical chromatogram derived from the fractionation of serum from one of the IgA-deficient patients with anti-IgA antibodies is shown in Fig.1. The presence of large complexes can be seen as a peak at the column void volume.
Fig. 1.
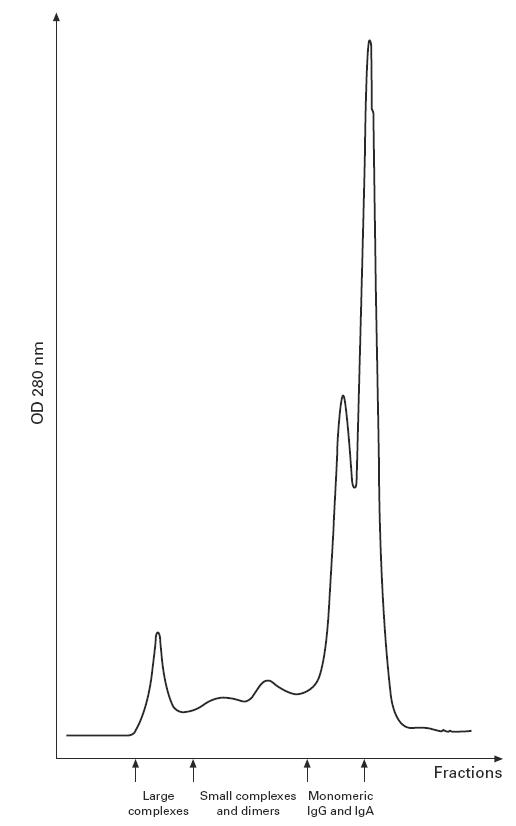
Chromatogram of an IgA-deficient serum with anti-IgA antibodies separated on a Superose 6 column. Fractions containing monomeric IgG, IgA, IgM and complexes are shown.
Serum samples taken before treatment and after disappearance of anti-IgA reactivity were fractionated and analysed for immunoglobulin content and anti-IgA activities in monomeric, dimeric/small complexes and large complex fractions. The anti-IgA activity detected in whole serum taken before treatment with IgA-containing immunoglobulin resided primarily in the monomeric fraction. After a period of immunoglobulin treatment, anti-IgA was no longer detectable in whole sera (Table 2), but could be recovered in the dimeric and complex fractions. Moreover, the IgA-containing fraction was shifted to the complex-containing part of the chromatogram (Figs 2 and 3a,b).
Fig. 2.
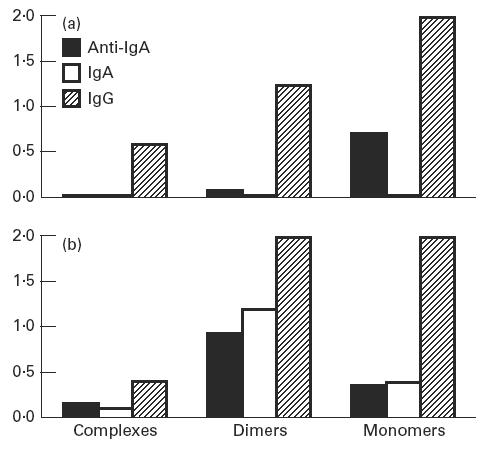
ELISA optical density values from measurement of IgG, IgA and anti-IgA activity in fractionated serum from patient 3. (a) Before treatment. (b) After 1 year of treatment with immunoglobulin containing < 3% IgA ( < 5 mg/ml).
Fig. 3.
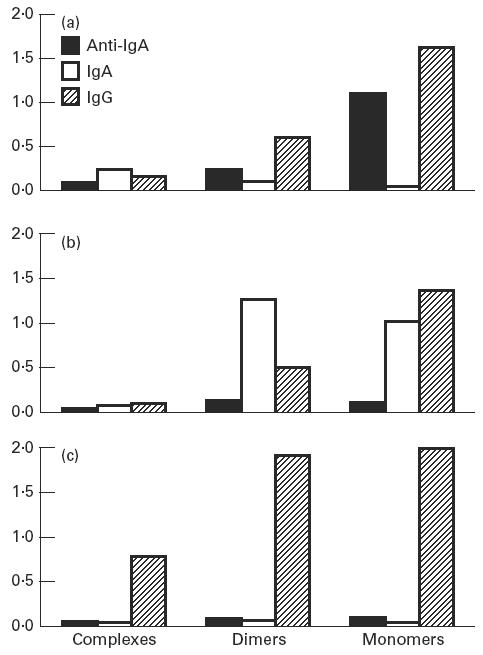
ELISA optical density values from measurements of IgG, IgA and anti-IgA activity in fractionated serum from patient 2. (a) Before treatment. (b) After 3 months of treatment with immunoglobulin containing < 3% IgA ( < 5 mg/ml). (c) After 1 year of treatment with immunoglobulin containing < 0.01% IgA ( < 80 μg/ml).
Samples from patient 2 receiving the low IgA preparation ( < 80 μg/ml) were negative for both IgA and anti-IgA in unfractionated and fractionated serum, but a large proportion of the total IgG was found in the complexes (Fig. 3c).
Addition of purified IgA to a serum containing high titres of anti-IgA
In vitro experiments where purified IgA was added to a serum containing IgG anti-IgA antibodies resulted in a redistribution of IgG, IgA and anti-IgA between fractions representing large complexes, dimers and monomers, as shown in Fig. 4. With no IgA added, IgG and anti-IgA were evenly distributed in monomer and dimer fractions. Some IgG could also be found in the large complex fraction (Fig. 4a). When 10 μg of IgA were added per millilitre of serum, the level of anti-IgA decreased in the monomeric fractions with a simultaneous increase in the large complex fraction. IgA could barely be detected in spite of the fact that the detection limit of the IgA ELISA is several orders of magnitude lower than the concentration added (10 μg/ml) (Fig. 4b). With increasing concentration of IgA added (100 μg to 1 mg of IgA/ml serum), the anti-IgA signal decreased to close to the detection limit while the IgA level increased, especially in the complex and dimer fractions. The IgG signal was simultaneously slightly increased in the complex fractions (Fig. 4c,d).
Fig. 4.
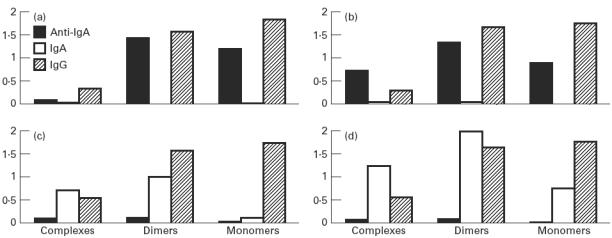
Addition of purified IgA to serum from a patient with IgA deficiency and anti-IgA. ELISA optical density values from measurements of IgG, IgA and anti-IgA activity after column fractionation. (a) No IgA added. (b) IgA 10 μg/ml serum. (c) IgA 100 μg/ml serum. (d) IgA 1 mg/ml serum.
DISCUSSION
The following interpretation of the results from the in vitro experiments can help in understanding the findings obtained with patient samples. With a low amount of IgA added, the IgG anti-IgA was capable of binding all of the IgA present, still retaining a proportion of free monomeric IgG anti-IgA. The complexes formed were both IgG-IgA dimers as well as larger complexes, but the amount of antigen (IgA) in the complexes was small enough to permit a favourable competition by the IgA coated onto the ELISA plate. Hence, IgG anti-IgA activity in the complex fractions is paradoxically detected in spite of their binding sites already being occupied.
When the IgA concentration was further increased, the stoichiometry of the complexes changed to a situation of antigen excess. Monomeric IgG-anti-IgA was no longer seen. The anti-IgA antibodies present in complexes were cross-bound and not available for binding to IgA in the ELISA plate and hence not detected. Under these conditions, it was possible to detect most of the IgA added, although most was bound in complexes with IgG.
With the above interpretation in mind we can explain the changes seen in immunoglobulin levels and anti-IgA activities in the patient samples by a simple mechanism. Some of the added IgA present in the immunoglobulin preparations will be bound by the anti-IgA already in the circulation. Complexes will be formed containing both IgG and IgA, and if the stoichiometry is favourable, anti-IgA activity.
When the serum level of IgA is increased, there is an excess of IgA (antigen) present that will competitively inhibit any reaction to the IgA coated to the ELISA plate. When the sera are fractionated and dimeric and complex fractions are analysed, monomeric IgA is no longer present, hence not available for competition. Anti-IgA activity can then be detected in these fractions. One of the patients (patient 2) does not fit into the concept, as we did not detect any IgA or anti-IgA in either whole serum or in any of the fractions, but there were appreciable amounts of IgG in all fractions. The explanation for the total disappearance of anti-IgA is not clear, but one possibility might be induction of tolerance by the long-term infusion of IgG and low levels of IgA. A possible mechanism for this is as follows.
The level of autoreactive antibodies in normal individuals is thought to be controlled by anti-idiotypic antibodies [14]. In diseases due to autoantibodies there are examples of cure/disappearance of the harmful autoantibodies due to spontaneous evolvement of anti-idiotypic antibodies [15–17]. There are reports of the disappearance of serum autoantibodies and several examples of apparent cure from a disease after high-dose immunoglobulin treatment [18]. Several hypotheses for the mechanisms causing these effects have been put forward, but they are as yet largely unproven (for reviews see Jungi & Nydegger [19] and Hammarström et al. [20]). One possibility for autoantibody suppression after IVIG is based on the finding that anti-idiotypic antibodies present in the immunoglobulin preparation can interact with the autoreactive antibodies and cause complex formation. The complexes could then be cross-linked by surface immunoglobulin and Fc-receptors on the surface of B cells, thereby inducing anergy [21]. For anti-IgA autoantibodies, the mechanism described above is attractive since the immunoglobulin preparation contains IgA, which would serve as autoantigen causing enhanced complex formation and, more importantly, direct specificity towards B cell clones producing anti-IgA.
Another alternative might be that the slow pace of IgG production (and anti-IgA) in the patient is exactly counterbalanced by the continuous supplementation of low levels of exogenous IgA, and the rate is so low that the complexes formed are cleared from the circulation.
The present study demonstrates a possibility of safely inducing unresponsiveness against IgA in immunodeficient patients with anti-IgA antibodies, by initial subcutaneous infusion with a high IgA-containing (up to 3%) immunoglobulin preparation. This might reduce the need for total IgA depletion of the immunoglobulin preparations for subcutaneous infusion. Immunoglobulin treatment of patients with ‘selective’ IgA deficiency is, in general, not recommended, mainly because of adverse reactions when IgA-containing immunoglobulin products have been given intravenously [5,8]. None of the four patients presented here showed any systemic side effects, nor any evidence of immune complex deposition, and only occasional, mild local effects at the site of infusion were noted. These side effects were not different from those in the group without anti-IgA antibodies.
The finding that immunoglobulin can be given to IgA-deficient patients without side effects when infused subcutaneously, and is clinically efficacious [22], suggests that this route of administration should be recommended for infection-prone IgA-deficient patients. It is, however, important to stress the need for a careful surveillance and monitoring of these patients when new therapeutic strategies are employed.
Acknowledgments
This work was supported by grants from the The Swedish Medical Research Council and from Pharmacia-Upjohn.
References
- 1.Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9:722–8. [PubMed] [Google Scholar]
- 2.Ferreira A, Garcia Rodriguez MC, Lopez-Trascasa M, et al. Anti-IgA antibodies in selective IgA deficiency and in primary immunodeficient patients treated with gammaglobulin. Clin Immunol Immunopathol. 1988;47:199–207. doi: 10.1016/0090-1229(88)90072-4. [DOI] [PubMed] [Google Scholar]
- 3.Björkander J, Hammarström L, Smith CIE, et al. Immunoglobulin prophylaxis in patients with antibody deficient syndromes and anti-IgA antibodies. J Clin Immunol. 1987;7:8–15. doi: 10.1007/BF00915419. [DOI] [PubMed] [Google Scholar]
- 4.Koskinen S, Tolo H, Hirvonen M, Koistinen J. Long term follow up of anti-IgA antibodies in healthy IgA-deficient adults. J Clin Immunol. 1995;15:194–8. doi: 10.1007/BF01541089. [DOI] [PubMed] [Google Scholar]
- 5.Burks AW, Sampson HA, Buckley RH. Anaphylactic reactions after gamma globulin administration in patients with hypogammaglobulinemia. Detection of IgE antibodies to IgA. N Engl J Med. 1986;314:560–4. doi: 10.1056/NEJM198602273140907. [DOI] [PubMed] [Google Scholar]
- 6.Frankel SJ, Polmar SH, Grumet FC, Wedner HJ. Anti-IgA antibody associated reactions to intravenous gammaglobulin in a patient who tolerated intramuscular gammaglobulin. Ann Allergy. 1986;56:436–9. [PubMed] [Google Scholar]
- 7.Martinez-Sanz R, Marsa L, De La Llana R, et al. Anaphylactic reaction associated with anti-IgA antibodies. Description of one case successfully treated by means of extracorporeal circulation. J Cardiovasc Surg. 1990;31:247–8. [PubMed] [Google Scholar]
- 8.Cunningham-Rundles C, Zhou Z, Mankarious S, Courter S. Long-term use of IgA depleted intravenous immunoglobulin in immunodeficient subjects with anti-IgA antibodies. J Clin Immunol. 1993;13:272–8. doi: 10.1007/BF00919386. [DOI] [PubMed] [Google Scholar]
- 9.Gardulf A, Hammarström L, Smith CIE. Home treatment of hypogammaglobulinaemia with subcutaneous gammaglobulin by rapid infusion. Lancet. 1991;338:162–6. doi: 10.1016/0140-6736(91)90147-h. [DOI] [PubMed] [Google Scholar]
- 10.Cunningham-Rundles C, Wong S, Björkander J, Hanson L-Å. Use of an IgA depleted intravenous immunoglobulin in a patient with an anti-IgA antibody. Clin Immunol Immunopathol. 1986;38:141–9. doi: 10.1016/0090-1229(86)90133-9. [DOI] [PubMed] [Google Scholar]
- 11.Baudner S, Bienvenu J, Blirup-Jensen S, et al. 1993. pp. 1–172. The certification of a matrix reference material for immunochemical measurement of 14 human serum proteins. CRM 470. Brussels: Community Bureau of the European Communities,
- 12.Hammarström L, Persson MAA, Smith CIE. Anti-IgA in selective IgA deficiency: in vitro effects and Ig subclass pattern of human anti-IgA. Scand J Immunol. 1983;18:8–15. doi: 10.1111/j.1365-3083.1983.tb00885.x. [DOI] [PubMed] [Google Scholar]
- 13.Hiemstra PS, Gorter A, Stuurman ME, et al. Activation of the alternative pathway of complement by human serum IgA. Eur J Immunol. 1987;17:321–6. doi: 10.1002/eji.1830170304. [DOI] [PubMed] [Google Scholar]
- 14.Jerne NK. Toward a network theory of immunity. Ann Immunol (Paris) 1991;125C:373–89. [PubMed] [Google Scholar]
- 15.Sultan Y, Rossi F, Kazatchkine MD. Recovery from anti-VIII.C (antihemophilic factor) autoimmune disease is dependent on generation of antiidiotypes against anti-VIII:C autoantibodies. Proc Natl Acad Sci USA. 1987;84:828–31. doi: 10.1073/pnas.84.3.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruiz-Arguelles A. Spontaneous reversal of acquired autoimmune dysfibrinogenemia probably due to an antiidiotypic antibody directed to an interspecies cross-reactive idiotype expressed on antifibrinogen antibodies. J Clin Invest. 1988;82:958–63. doi: 10.1172/JCI113704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossi F, Jayne DR, Lockwood CM, Kazatchkine MD. Anti-idiotypes against anti-neutrophil cytoplasmic antigen autoantibodies in normal human polyspecific IgG for therapeutic use in the remission sera of patients with systemic vasculitis. Clin Exp Immunol. 1991;83:298–303. doi: 10.1111/j.1365-2249.1991.tb05631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bussel JB, Goldman A, Imbach P, et al. Treatment of acute idiopathic thrombocytopenia of childhood with intravenous infusions of gammaglobulin. J Pediatrics. 1985;106:886–90. doi: 10.1016/s0022-3476(85)80231-6. [DOI] [PubMed] [Google Scholar]
- 19.Jungi TW, Nydegger UE. Proposed mechanisms of action of intravenous IgG (IVIG) in autoimmune diseases. Transfus Sci. 1992;13:267–90. [Google Scholar]
- 20.Hammarström L, Gardulf A, Hammarström V, et al. Systemic and topical immunoglobulin treatment in immunocompromised patients. Immunol Rev. 1994;139:43–70. doi: 10.1111/j.1600-065x.1994.tb00856.x. [DOI] [PubMed] [Google Scholar]
- 21.Phillips NE, Parker DC. Cross-linking of B lymphocyte Fc gamma receptors and membrane immunoglobulin inhibits anti-immunoglobulin-induced blastogenesis. J Immunol. 1984;132:627–32. [PubMed] [Google Scholar]
- 22.Gustafson R, Gardulf A, Granert C, et al. Prophylactic therapy for selective IgA deficiency. Lancet. 1997;350:865. doi: 10.1016/S0140-6736(05)62034-X. [DOI] [PubMed] [Google Scholar]