

Abstract
It is known that antiphospholipid antibodies (aPL) hamper the anticoagulant activity of the protein C system, but the mechanism is still obscure. In this study, we demonstrate that anticardiolipin antibodies (not anti-protein C autoantibodies) can bind protein C via β2-GPI, which bears their binding epitope, in a fashion dependent on negatively charged phospholipids. We studied the binding of IgG from aPL to protein C in the presence of β2-GPI by ELISA (anti-‘protein C’ antibody ELISA), and compared their binding with those obtained in the absence of β2-GPI. In the anti-‘protein C’ antibody ELISA system, 47% of 78 aPL+ patients had a positive titre in the presence of cardiolipin (CL) and β2-GPI, but binding was not found in the absence of β2-GPI. Highly significant correlations were found between the titre of anti-‘protein C’ antibody in the presence of β2-GPI and that of anti-β2-GPI antibody (r = 0.802, P = 0.0001). We further analysed the interaction between protein C, phospholipids, β2-GPI and human aCL MoAbs established from patients with antiphospholipid syndrome. In a first set of experiments, the binding of β2-GPI to protein C and its phospholipid dependency were investigated. β2-GPI bound to protein C in the presence of CL or phosphatidylserine, but not in the presence of phosphatidylcholine or phosphatidylethanolamine. In a second group of experiments, the binding of three human monoclonal aCL recognizing the cryptic epitope of β2-GPI (virtually anti-β2-GPI antibodies) was evaluated in the presence of cardiolipin and β2-GPI. All three human monoclonal aCL bound to protein C in the presence of CL and β2-GPI, whereas they did not in the absence of either β2-GPI or CL. These data suggest that protein C could be a target of aCL by making a complex with CL and β2-GPI, leading to protein C dysfunction.
Keywords: thrombosis, antiphospholipid syndrome, coagulation, phospholipid-binding protein
INTRODUCTION
Antiphospholipid antibodies (aPL) are being recognized as a heterogeneous group of antibodies whose specificity is directed not only towards phospholipids but also towards phospholipid-binding proteins or phospholipid–protein complexes [1,2]. The presence of aPL is associated with arterial and venous thrombosis, recurrent fetal loss, neurological disorders, pulmonary hypertension and thrombocytopenia. The term ‘antiphospholipid syndrome’ (APS) was coined to link these clinical manifestations with the persistence of aPL, which is now recognized as one of the most common causes of acquired thrombophilia [3,4]. In 1990, three groups reported that anticardiolipin antibodies (aCL) bound to cardiolipin (CL) in the presence of an ‘aCL cofactor’, namely β2-GPI [5–7]. β2-GPI is a 50-kD phospholipid-binding protein with a plasma concentration of about 200 μg/ml [8]. It is formed by five short consensus repeat domains found also in various proteins of mammalian origin [9,10]. The phospholipid-binding site is present within the fifth domain of β2-GPI [11], whereas the possible epitope for aCL binding seems to be located in the fourth domain [12]. Matsuura et al. [13] and Roubey et al. [14] showed that aCL recognized this epitope in the absence of CL if β2-GPI was coated onto polystyrene plates where oxygen was introduced by radiation, implying that aCL can bind not only CL–β2-GPI complex but also to β2-GPI alone. Furthermore, it has been shown that anti-β2-GPI antibodies detected by this irradiated plate system are associated with clinical features of APS [15,16].
Since β2-GPI has strong binding properties to negatively charged proteins or phospholipids involved in coagulation processes, it is likely that aPL may hamper β2-GPI-associated coagulation steps. β2-GPI inhibits ADP-induced platelet aggregation [17] and factor XII activation [18], therefore the intrinsic clotting pathway [19] and factor Xa generation [20]. Despite these regulatory functions, familial deficiency of β2-GPI is not a risk factor for thrombosis [21], and decreased levels are not usually associated with APS [22]. Therefore β2-GPI deficiency alone cannot account for thrombosis, but it might play a critical role in a multifactorial event.
The protein C system is one of the most important anti-thrombotic pathways mediated by the vessel wall [23]. Thrombin immobilized by thrombomodulin on endothelial cells cannot activate platelets, nor convert fibrinogen into fibrin, but may still activate protein C [24,25]. Activated protein C (APC) further inhibits thrombin generation by proteolytic inactivation of factor Va and VIIIa in the presence of protein S [26,27]. Subjects with familial protein C deficiency or with APC resistance, a congenital abnormality where APC cannot inactivate factor Va because of a mutation of the latter, are exposed to an increased risk of thrombosis [28–30].
Several studies have demonstrated an interaction between aPL and the protein C system [31]. A number of publications have shown that IgG purified from aPL+ sera inhibit the activity of protein C [32–35]. However, β2-GPI, one of the most important targets of aPL, also shows an inhibitory effect on both protein C activation by thrombin/thrombomodulin [36] and APC activity on factor Va degradation [37]. These effects of aPL on the protein C pathway cannot be explained solely by a dysfunction of β2-GPI in the affected patients. Some investigators have suggested the presence of autoantibodies against protein C or against protein C–phospholipid complex [35], but these antibodies were detectable only under certain experimental conditions and their clinical significance, such as an association with acquired protein C deficiency, sometimes found in APS patients [38], has not been established.
In this study, we demonstrate that aPL+ IgG may bind to protein C in the presence of both CL and β2-GPI. Binding activities were strongly correlated with anti-β2-GPI antibody titres. To clarify the interaction between protein C, phospholipid, β2-GPI and aPL, we employed three human monoclonal aCL raised from patients with APS, which bound to β2-GPI–anionic phospholipid complex and to β2-GPI coated on oxygen-induced plastic plates (virtually anti-β2-GPI antibodies). These properties are representative of the majority of aCL associated with APS.
PATIENTS AND METHODS
Patients
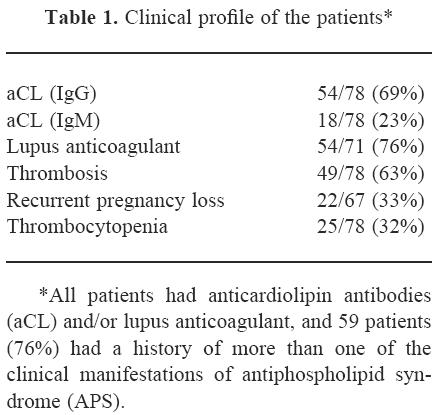
The study population comprised 78 aPL (aCL and/or LA) positive patients (67 female, 11 male; mean age 42 years, range 12–66 years), 32 patients (41%) with primary APS, 27 (35%) with APS secondary to systemic lupus erythematosus (SLE) and 19 (24%) with SLE without APS, who were selected randomly from our APS or SLE patient data base, and their clinical records were carefully reviewed retrospectively. All patients fulfilled the proposed criteria for APS and/or the American College of Rheumatology criteria for the classification of SLE. Clinical profiles of these patients are given in Table 1.
Table 1.
Clinical profile of the patients *
Anti-‘protein C’ antibody ELISA
Microtitre plates, Immulon 4 (Dynatech Labs, Inc., VA) were coated with 2.5 U/ml of human protein C (CRST, Lille, France) in Tris-buffered saline (TBS) at 4°C and washed twice in TBS. To avoid non-specific binding of proteins, wells were blocked with 150 μl of TBS containing 10% bovine serum albumin (BSA, A-7906; Sigma Chemical Co., St Louis, MO). After three washes in TBS containing 0.05% Tween 20 (Sigma) (TBS–T), 50 μl of serum diluted 1:200 with TBS containing 1% BSA with 0.1 mm of CaCl2 (BSA–Ca) and 30 μg/ml of cardiolipin liposome in the presence or absence of 20 μg/ml of human β2-GPI were added in duplicate (human β2-GPI purified from normal human sera by sequential CL-affinity, ion exchange column and protein A-Sepharose column chromatography [39] was a generous gift from the Immunology Laboratory, Diagnostics Division, Yamasa Corp., Japan). Plates were incubated for 3 h at room temperature and washed three times with TBS–T. Fifty microlitres/well of the appropriate dilution of alkaline phosphatase-conjugated goat anti-human IgG (Sigma) in TBS containing 1% BSA were added. After 1 h of incubation at room temperature and after four washes in TBS–T, 100 μl/well of 1 mg/ml p-nitrophenylphosphate disodium (Sigma) in 1 m diethanolamine buffer pH 9.8 were added. Following colour development, optical density at 405 nm (OD405) was measured by a Titertek Multiskan MC apparatus (Flow Labs, Herts, UK).
The assay was repeated using the IgG fraction of sera from two patients showing high (IgG (A)) and low (IgG (B)) β2-GPI binding according to the ELISA described in the following paragraph. Different concentrations of these purified IgG (20, 10, 5 and 2.5 μg/ml) substituted for patients' sera, and IgG binding was evaluated in the same manner.
Anti-β2-GPI antibody ELISA
Anti-β2-GPI was detected by ELISA utilizing irradiated ELISA plates as described previously [16]. Briefly, irradiated microtitre plates (Sumilon type C; Sumitomo Bakelite, Tokyo, Japan) were coated with purified human β2-GPI in PBS at 4°C overnight. Wells were blocked with 3% gelatin for 1 h at 37°C. After three washes with PBS–T, 50 μl of serum diluted in PBS containing 1% BSA in 1:50 were added in duplicate. Plates were incubated for 1 h at room temperature, followed by alkaline phosphatase-conjugated goat anti-human IgG and substrate. Anti-β2-GPI titre of each sample was derived from the standard curve according to the dilutions of the positive control.
Binding of protein C to phospholipids
To clarify the mechanism of the binding of IgG, we first evaluated the binding of protein C to phospholipids. Fifty microlitres/well of 150 μg/ml CL in ethanol or phosphatidylserine (PS), phosphatidylcholine (PC) and phosphatidylethanolamine (PE) in 1:4 chloroform/methanol were added to the microtitre plates, Immulon I (Dynatech Labs). After evaporation of the solvent, plates were blocked with 150 μl of TBS containing 1% BSA. Plates were then washed three times with TBS and 50 μl of 12.5 U/ml protein C in BSA–Ca were added, followed by 2 h incubation at room temperature. Alkaline phosphatase-conjugated polyclonal rabbit anti-human protein C antibody (50 μl; Sigma) in TBS–BSA was added after three washes with TBS, and the plates were incubated for 1 h at room temperature. After four washes with TBS, 100 μl/well of the substrate were added and OD405 was measured in the same way.
Binding of β2-GPI to protein C in the presence of phospholipids
The binding of β2-GPI to protein C was tested in the presence of different phospholipids. Protein C-coated plates were prepared and blocked as described in the ‘Anti-‘protein C’ antibody ELISA’ section. Fifty microlitres of 5 μg/ml β2-GPI in BSA–Ca were added in the presence of 30 μg/ml CL, PS, PC or PE, and the plates were incubated for 2 h at room temperature. Bound β2-GPI was detected by rabbit polyclonal anti-human β2-GPI sera (Diagnostica Stago, Asnieres, France), followed by ALP-conjugated anti-rabbit immunoglobulin and the substrate. The binding between β2-GPI and protein C was also tested by adding different concentrations of biotinylated β2-GPI, which had been prepared with a standard biotinylation protocol (Clontech, Palo Alto, CA), to the same system in the presence/absence of CL, followed by ALP-conjugated avidin (Sigma) and the substrate.
Human monoclonal aCL
The establishment of human monoclonal aCL has been described in detail elsewhere [40]. Briefly, peripheral blood lymphocytes were obtained from patients with APS. Isolated B cells were transformed with Epstein–Barr virus, and were fused with mouse–human heterohybridoma cell line (SHM-D33) (no 1668-CRL; American Type Culture Collection, Rockville, MD) according to a standardized methodology after 3–4 weeks of culture. Cells producing aCL were detected by a conventional aCL ELISA. Cloned hybridomas were finally cultured in serum-free medium (EX-Cell 300; Sera-Lab). MoAbs were precipitated from culture supernatant using 50% saturated ammonium sulphate, then filtrated on PD-10 Columns (Pharmacia LKB Biotechnology).
Three human IgM monoclonal aCL (EY2C9, EY1C8 and GR1D5) and one control monoclonal IgM lacking aCL activity (TM1B9) were used in this study.
The binding of human monoclonal aCL to protein C
The binding between different concentrations of monoclonal aCL (10, 5, 2.5, 1.25, 0.63 μg/ml) and protein C was investigated by the anti-‘protein C’ antibody ELISA, in which ALP-conjugated goat anti-human IgG was substituted with ALP-conjugated mouse anti-human IgM MoAb. This assay was repeated with 10 μg/ml of EY2C9 and GR1D5 in the presence/absence of β2-GPI (20 μg/ml) and CL (30 μg/ml).
Inhibition assay
Two high binding serum samples and EY2C9 (monoclonal aCL) were used in the inhibition assay. Serum samples or EY2C9 were preincubated with different concentrations of protein C (3.1–25 U/ml) at 4°C overnight, and anti-‘protein C’ antibody ELISA was performed on these samples. The same concentrations of human prothrombin (Enzyme Research Laboratory, Swansea, UK) were used as a control inhibitor.
The effect of monoclonal aCL (EY2C9) on the protein C system
The effect of monoclonal aCL on the protein C system was tested by a clotting assay. Different concentrations of EY2C9 or TM1B9 in TBS–BSA were added to 90% normal plasma, and activated partial thromboplastin time (APTT) was measured in the presence/absence of APC utilizing a commercial kit (Coatest APC Resistance; Chromogenix AB, Sweden). ‘Activated protein C resistance phenomenon’ was expressed as the ratio (APTT with activated protein C/APTT without).
Statistical analysis
The statistical analysis was performed by Statview II (Apple Macintosh software). The values of the anti-‘protein C’ antibody were compared between two groups by the Mann–Whitney non-parametric test.
RESULTS
Anti-‘protein C’ antibody and its β2-GPI dependency
Thirty-seven (47%) aPL+ IgG bound to protein C in the presence of β2-GPI, but only one (4%) of 23 healthy controls did. Without β2-GPI, OD405 were markedly lower than those with β2-GPI (Fig. 1a). Among the aPL+ sera, OD values of anti-‘protein C’ antibody in the presence of β2-GPI (anti-‘protein C’ antibody/β2-GPI) in patients with previous history of thrombosis (n = 49) were significantly higher than those of patients without a history of thrombosis (n = 29) (Fig. 1b).
Fig. 1.

Binding of antiphospholipid syndrome (APS) patient IgG to protein C. (a) Diluted serum samples were added to protein C-coated plates with cardiolipin (CL) in the presence (anti-‘protein C’ antibody/β2-GPI) or absence of β2-GPI. In the presence of β2-GPI, 37 serum samples showed positive bindings, but in most samples, binding values were markedly reduced in the absence of β2-GPI. Horizontal line represents the mean + 2 s.d. of 23 healthy controls. (b) Association between anti-‘protein C’ antibody/β2-GPI and a history of thrombosis. In this series, patients with a history of thrombosis showed higher binding to protein C in the presence of β2-GPI than those without history of thrombotic events.
Anti-β2-GPI antibody was detected in 40 (51%) patients. A highly significant correlation was found between the titres of anti-‘protein C’ antibody/β2-GPI and those of anti-β2-GPI antibody, more than the correlation between the former and the titres of the standard aCL (Fig. 2).
Fig. 2.
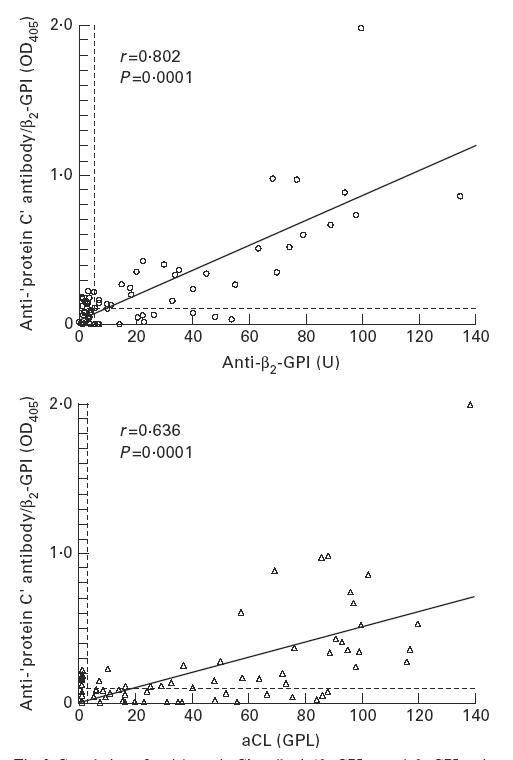
Correlation of anti-‘protein C’ antibody/β2-GPI to anti-β2-GPI antibody and to conventional anticardiolipin antibodies (aCL). Anti-β2-GPI antibody titre was determined by ELISA utilizing irradiated ELISA plates. Anti-‘protein C’ antibody/β2-GPI titres correlated both to anti-β2-GPI antibody and to aCL titres; the former correlation was greater than the latter.
IgG fraction binding to protein C and its β2-GPI dependency
IgG (A) which bound β2-GPI on irradiated ELISA plate (data not shown) showed protein C binding in a dose-dependent manner only in the presence of β2-GPI with CL. However, IgG (B), lacking β2-GPI binding, showed little binding to protein C both in presence and absence of β2-GPI (Fig. 3).
Fig. 3.
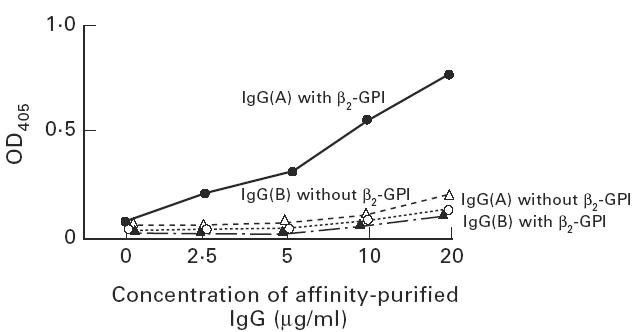
Binding of purified IgG to protein C. IgG fractions were prepared from two patients with antiphospholipid antibodies (aPL). IgG (A) showed the binding to β2-GPI coated on irradiated plates, but IgG (B) did not (data not shown). Both IgG were added to protein C-coated plate with cardiolipin (CL) in the presence/absence of β2-GPI. IgG (A) with β2-GPI bound to protein C, but IgG (A) without β2-GPI, IgG (B) with/without β2-GPI showed little binding.
Binding between phospholipids and protein C, and between protein C and β2-GPI in the presence of phospholipids
Protein C bound to all four phospholipids tested in these experimental conditions. β2-GPI, however, bound protein C only in the presence of anionic phospholipids (CL and PS) (Fig. 4a). The binding between protein C and β2-GPI with CL was confirmed by another experiment, in which biotinylated β2-GPI showed binding to protein C only in the presence of CL (Fig. 4b).
Fig. 4.
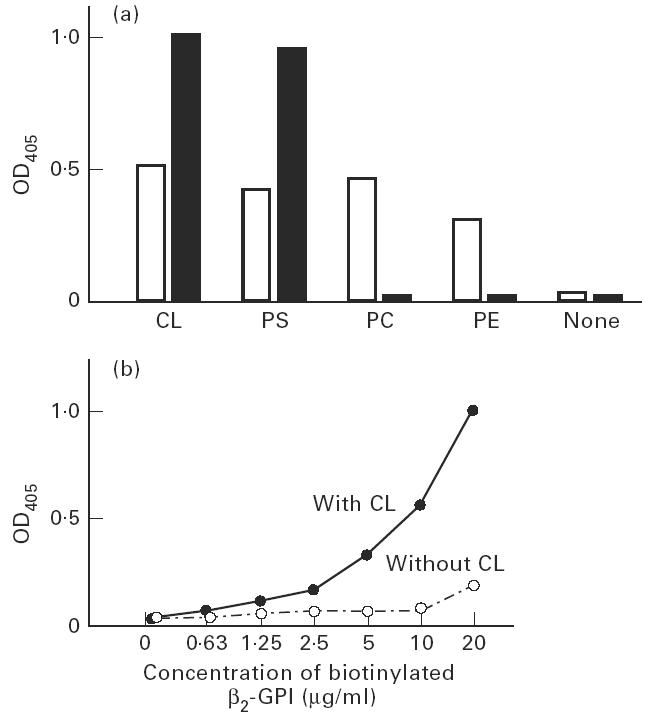
Binding of protein C, phospholipids and β2-GPI. (a) Binding of protein C to phospholipids coated on the plates (□), and binding of β2-GPI to protein C (▪) in the presence of phospholipids. Protein C bound to coated cardiolipin (CL), phosphatidylserine (PS), phosphatidylcholine (PC) and phosphatidylethanolamine (PE). However, the binding of β2-GPI to protein C depended on the presence of anionic phospholipids (CL and PS) but not neutral phospholipids (PC and PE). Bound protein C and bound β2-GPI were detected by rabbit polyclonal anti-protein C antisera or anti-β2-GPI antisera, respectively. (b) Binding of biotinylated β2-GPI to protein C. Biotinylated β2-GPI bound to protein C in the presence of CL, but not in its absence.
Binding of human monoclonal aCL to protein C
The characteristics and specificity of these monoclonal aCL were described previously [40]. In summary, all monoclonal aCL that were used in this experiment (EY2C9, EY1C8 and GR1D5) were IgM and had aCL activity in the presence of calf serum containing bovine β2-GPI, or of purified human β2-GPI. They also bound to other anionic phospholipids in the same condition. In the absence of β2-GPI, however, they did not bind to phospholipids. Furthermore, they bound β2-GPI coated on irradiated ELISA plates in the absence of CL, but did not bind β2-GPI coated on plain ELISA plates, nor β2-GPI in a liquid phase.
All three monoclonal aCL bound to protein C in the presence of β2-GPI/CL in a dose-dependent manner, but control monoclonal IgM did not (Fig. 5a). Ten micrograms/ml of EY2C9 and GR1D5 bound protein C in the combined presence of β2-GPI and CL, but did not bind protein C in the presence of β2-GPI alone or CL alone (Fig. 5b).
Fig. 5.
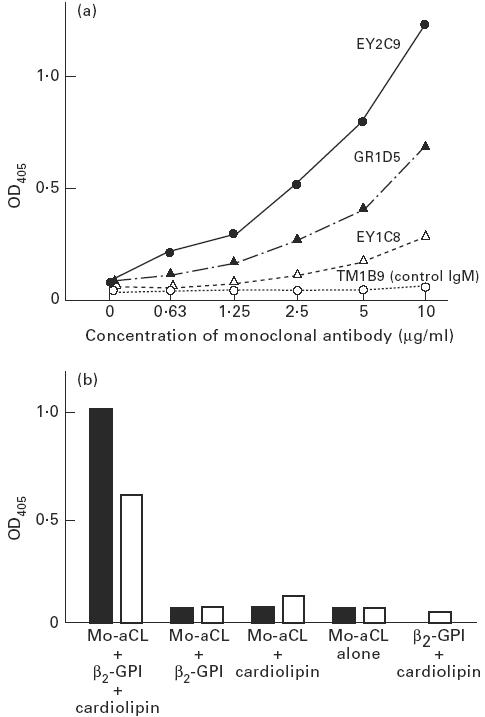
Binding of monoclonal anticardiolipin antibodies (aCL) to protein C. (a) All three human monoclonal aCL (EY2C9, GR1D5 and EY1C8) bound to protein C coated on the plates in the presence of CL and β2-GPI, but TM1B9 (control human monoclonal IgM) lacking aCL activity did not. (b) Monoclonal aCL (▪, EY2C9; □, GR1D5) bound to protein C only in the presence of a combination of CL and β2-GPI. In the absence of CL and/or β2-GPI, little binding of monoclonal aCL to protein C was found.
In all experiments, the bindings of monoclonal aCL to negative control wells (i.e. bindings to blocked plates without protein C) were always negligible (OD405 < 0.055).
Inhibitory effect of liquid-phase protein C
The inhibition assay was performed using two high binding sera and EY2C9. Liquid-phase protein C or prothrombin were added as inhibitors, and the binding was inhibited by protein C but not by prothrombin (Fig. 6).
Fig. 6.
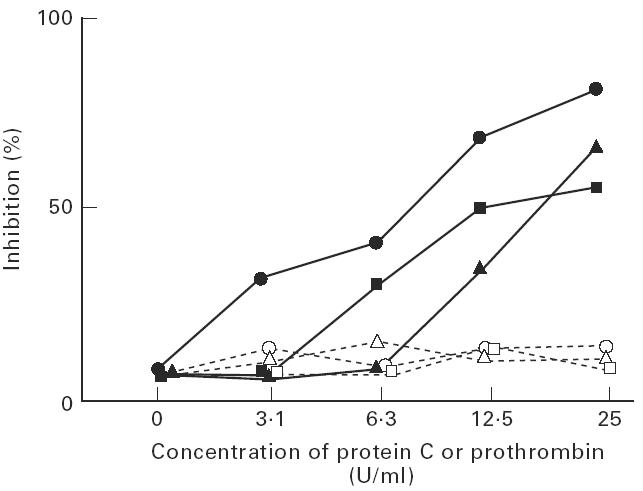
Inhibition of the binding of IgG or EY2C9 to protein C by excess of liquid phase protein C. Anti-‘protein C’ antibody ELISA was performed in two serum samples (•, ▴) or EY2C9 (▪) in the presence of cardiolipin (CL) and β2-GPI after incubating with different concentrations of protein C (closed symbols) or prothrombin (open symbols). The binding was inhibited by protein C but not by prothrombin.
The effect of monoclonal aCL on the protein C system
One of monoclonal aCL, showing the highest binding to protein C, was investigated for its effect on the protein C system. ‘APC resistance’ was tested in a normal plasma containing different concentrations of EY2C9 or TM1B9. High concentrations of EY2C9 markedly reduced the ratio (APTT with APC/APTT without), but the ratio remained high in a plasma containing the control monoclonal IgM (Fig. 7), implying that EY2C9 inhibited the function of APC leading to the ‘APC resistance phenomenon’.
Fig. 7.
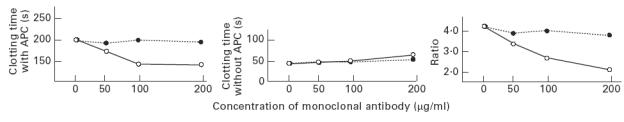
The effect of monoclonal anticardiolipin antibodies (aCL) on the protein C system. Different concentrations of EY2C9 (monoclonal aCL) and TM1B9 (control monoclonal IgM) were added to a normal plasma, and the activated partial thromboplastin time (APTT) with/without activated protein C (APC) was measured. The effects of APC in the plasma containing EY2C9 (○) and TM1B9 (•) were represented as a ratio (APTT with APC/APTT without). The prolongation of APTT by APC was reduced (expressed by the reduced ratio) in a plasma containing EY2C9 in a dose-dependent fashion.
DISCUSSION
All three human monoclonal aCL bound to protein C only in the presence of CL and β2-GPI, suggesting that aCL may bind to protein C, which traps CL and β2-GPI on the ELISA plates. This might imply that aCL can affect protein C via anionic phospholipids and β2-GPI, leading to a dysfunctional protein C pathway.
Those monoclonals are of IgM isotype. In general, IgM autoantibodies are not driven by specific antigen, but previous papers have shown the specificity and the characteristics of those monoclonal aCL [12,40,41]. Polyclonal IgG aCL prepared from APS patients competed with those monoclonal IgM for β2-GPI binding [40]. Recently it has been shown that those monoclonals activate cultured endothelial cells in a β2-GPI-dependent fashion as polyclonal IgG aCL do [41]. Thus, those monoclonals may represent the majority of aCL ( = anti-β2-GPI). As far as we have investigated, those monoclonals show no cross-reactivity against other antigens, such as negatively charged phospholipids, protein S (in the absence of β2-GPI), prothrombin, anti-thrombin III and heparan sulphate.
Since the first data regarding a correlation between aPL and the protein C system in 1983 [42], many studies have demonstrated their interactions. The fact that protein C and its cofactor protein S are phospholipid-binding plasma proteins has made this system one of the most likely to be involved in the pathophysiology of thrombosis in APS. APL may inhibit phospholipid-dependent reactions of the protein C pathway in different ways. First, they can interfere with the activation of protein C by the thrombin–thrombomodulin complex. Thrombin formation inhibited by aPL could paradoxically cause a prothrombotic tendency due to insufficient protein C activation [31,43]. It has also been shown that LA+ IgG inhibit the activation of purified protein C by thrombin on endothelial cells [44,45], or by thrombin and purified thrombomodulin [46]. Other studies, however, failed to confirm this phenomenon [36,47], possibly due to different experimental conditions such as the available amount of thrombomodulin expressed on cell surface and components of phospholipids present in the system. Second, the proteolytic effect of APC on factor Va/factor VIIIa can be inhibited by aPL. Several studies have supported this hypothesis. Marciniak & Romond [32] reported that factor Va degradation was reduced in plasma from patients with LA. Malia et al. [33] confirmed the inhibitory effect of IgG purified from aPL+ patients on factor Va degradation by APC, regardless of the presence of protein S in some of such plasma samples, and Borrell et al. [34] showed that purified IgG/M from patients with aPL disturb the anti-coagulant activity of APC on cultured human endothelial cells. Finally, the cofactor effect of protein S in the protein C pathway can be affected by aPL. A number of papers have shown a decreased plasma level of free protein S, the active form of protein S in patients with APS [48–50]. Oosting et al. reported that some of the IgG which inhibited factor Va degradation were directed not only against phospholipid-bound protein C but also against phospholipid-bound protein S [35].
According to the concept that aPL are directed not against phospholipids but against phospholipid-binding proteins or their complex with phospholipids [1], the most plausible concept is the presence of anti-protein C or antiphospholipid-protein C complex autoantibodies. Mitchell et al. [51] described a patient with massive thrombosis who presented an acquired protein C deficiency. Although they failed to prove a direct recognition of protein C by the patient's antibodies with Western blotting or immunoprecipitation, the IgG fraction of the plasma inhibited the functional anti-coagulant activity of APC, suggesting the presence of anti-protein C as an inhibitor. Oosting et al. [35] showed that the inhibitory effect of IgG on the protein C system was adsorbed by CL vesicles bound to protein C in some aPL+ plasmas, implying some of the IgG were directed to protein C–phospholipid complex. Ruiz-Argüelles et al. [52] showed the protein C binding of immunoglobulin in some patients with SLE by ELISA, but they could not correlate the presence of this antibody to protein C deficiency, nor to thrombotic events.
Initially, we tested the IgG binding from our serum samples to protein C in the presence of CL liposome by ELISA. Although some IgG bound to protein C, the binding values of aPL+ patients' samples were not impressive compared with those of healthy controls, and the number of high binding IgG was quite small. However, when purified β2-GPI was added to the sample diluent, the binding of IgG was markedly increased. In this system, sample sera contained small amounts of endogenous β2-GPI (≈ 1 μg/ml). When the binding of purified IgG was tested, the β2-GPI dependency of this binding was confirmed. Furthermore, the binding values to protein C strongly correlated with those of anti-β2-GPI titres on irradiated ELISA plates, suggesting the association between antibodies binding to protein C and aCL ( = anti-β2-GPI antibodies). Our experiments with human monoclonal aCL, whose binding epitopes exist on β2-GPI, confirmed the binding of aCL to protein C in the presence of CL and β2-GPI. Since the bindings of monoclonal aCL to negative control wells (blocked wells without protein C) were negligible, it was not possible that aCL bound to wells exposed to CL and β2-GPI without the interaction of protein C in these experiments. The binding was inhibited by excess of liquid-phase protein C, solidifying the (indirect) binding to protein C. There are two possible interpretations of these results. The first is that CL/β2-GPI forms a complex with protein C and aCL recognizes the epitope on β2-GPI bound to CL and protein C. The second is that aCL recognizes CL/β2-GPI in liquid phase and subsequently the CL–β2-GPI–aCL complex binds to protein C. The former is more likely, because the binding persisted when (i) CL, (ii) β2-GPI and (iii) aCL were added to protein C-coated plates independently in this order (the plates were washed after each step), but binding was lost if the sequence was changed to (i) β2-GPI, (ii) CL and (iii) aCL (data not shown). In any case, these data suggest that aCL could make a complex with protein C–CL–β2-GPI.
Protein C requires phospholipids to function, and this effect was believed to depend on anionic phospholipids such as PS or CL [53,54]. Recently it was reported that PE had a stronger effect on this system than PS or CL [55]. Our data showed that protein C bound not only to PS or CL but also to PC or PE, in keeping with the reported multiple phospholipid dependency of protein C function.
β2-GPI bound to protein C in the presence of CL or PS, shown by polyclonal anti-human β2-GPI sera as a detector of bound β2-GPI, and confirmed by biotinylated β2-GPI. In this binding between protein C and β2-GPI, CL or PS might have a ‘bridging’ role. On the other hand, this binding did not occur in the presence of PE or PC, nor in the absence of phospholipids. β2-GPI binds to anionic phospholipids but not to neutral ones, thus β2-GPI does not bind to protein C–PE nor to protein C–PC complexes. β2-GPI was reported to inhibit protein C activation by thrombin/thrombomodulin complex with CL vesicles [36], although a negligible effect was found on protein C activation on endothelial cells [47]. This discrepancy may be explained by the heterogeneous phospholipid dependency of protein C activation [31]. Furthermore, it was reported that β2-GPI inhibited the APC function on factor Va, which might occur by competitive inhibition of β2-GPI in the binding of APC to the phospholipid surface [37]. Therefore the effect of β2-GPI on protein C may be complex and heterogeneous. On the other hand, when phospholipid/β2-GPI binds to protein C and exposes the epitope for aCL, regardless of the function of β2-GPI on protein C, aCL could bind to β2-GPI accompanied with protein C, which might lead to protein C dysfunction. Our study clearly explains the paradox why both β2-GPI and anti-β2-GPI antibodies can have inhibitory effects on the protein C system.
However, aCL did not show the binding to prothrombin, another phospholipid-binding protein, in the presence of phospholipid/β2-GPI (data not shown), and prothrombin did not inhibit the binding of aCL to protein C/phospholipid/β2-GPI (Fig. 6). This inconsistency may be due to the differences in the way phospholipids interact with protein C and prothrombin, and further studies may be needed to clarify this characteristic behaviour of protein C.
Since several studies have already shown the effect of polyclonal antibodies containing aPL on the protein C system, we showed the effect of EY2C9, a monoclonal aCL used in this study, only in a crude functional experimental system. We found that normal plasma containing EY2C9 shows a reduced prolongation of APTT by APC, mimicking in an acquired fashion the congenital resistance of factor V to APC inactivation, due to the mutation in factor V (factor V:R506Q).
Despite considerable studies on the function of β2-GPI in vitro, the physiological role of β2-GPI is still uncertain. However, at least in the pathophysiology of the vascular events of APS, β2-GPI might play a role in binding cells or functional proteins with anionic phospholipids, thereby stimulating exposure of the epitope for aCL. In this case, irrespective of the function of β2-GPI itself, aCL might affect these β2-GPI-bound cells or proteins. IgG containing aCL bound to cultured endothelial cells in the presence of β2-GPI, and induced cell adhesion molecules such as E-selectin, vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) [56], which may lead to a procoagulant state. It is also known that aPL induce procoagulant substances such as tissue factor [57,58], plasminogen activator inhibitor-1 [59] and thromboxane A2 [60], in which processes aPL are likely to affect cells via β2-GPI or other phospholipid-binding proteins [61]. In terms of the binding mechanism of aPL, the binding of aCL to protein C with a combination of phospholipids and β2-GPI might be a similar phenomenon.
Recently Smirnov et al. [62] reported the crucial role of PE in the inhibitory effect of aPL on activated protein C activity. Our data show a second mechanism, and may in part explain the protein C dysfunction seen in patients with aCL+ APS.
Acknowledgments
The authors thank Immunology Laboratory, Diagnostics Division, Yamasa Corporation for providing human β2-GPI, and Dr Paul R. V. Ames (Haematology Department, St Thomas' Hospital, London) for valuable help. This work was partly supported by Lupus UK and Fondo de investigationes sanitarias, Spain, F.I.S. 95/5011, 96/5093.
References
- 1.Roubey RAS. Autoantibodies to phospholipid-binding plasma proteins: a new view of lupus anticoagulants and other ‘antiphospholipid’ antibodies. Blood. 1994;84:2854–67. [PubMed] [Google Scholar]
- 2.Roubey RAS. Immunology of the antiphospholipid antibody syndrome. Arthritis Rheum. 1996;39:1444–54. doi: 10.1002/art.1780390903. [DOI] [PubMed] [Google Scholar]
- 3.Hughes GRV. The antiphospholipid syndrome: ten years on. Lancet. 1993;342:341–4. doi: 10.1016/0140-6736(93)91477-4. [DOI] [PubMed] [Google Scholar]
- 4.Khamashta MA, Hughes GRV. Antiphospholipid antibodies and antiphospholipid syndrome. Curr Opin Rheumatol. 1995;7:389–94. doi: 10.1097/00002281-199509000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Galli M, Comfurius P, Maassen C, et al. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet. 1990;335:952–3. doi: 10.1016/0140-6736(90)91374-j. [DOI] [PubMed] [Google Scholar]
- 6.Matsuura E, Igarashi Y, Fujimoto M, et al. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet. 1990;336:177–8. doi: 10.1016/0140-6736(90)91697-9. [DOI] [PubMed] [Google Scholar]
- 7.McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti-phospholipid antibodies are directed against a complex antigen that induces a lipid-binding inhibitor of coagulation: β2-glycoprotein I (apolipoprotein H) Proc Natl Acad Sci USA. 1990;87:4120–4. doi: 10.1073/pnas.87.11.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato H, Enjyoji K. The presence of five Sushi domains. Biochem. 1991;30:11687–94. doi: 10.1021/bi00114a012. Amino acid sequence and location of the disulfide bonds in bovine β2-glycoprotein I. [DOI] [PubMed] [Google Scholar]
- 9.Ichinose A, Bottenus RE, Davie EW. Structure of transglutaminase. J Biol Chem. 1990;265:13411–4. [PubMed] [Google Scholar]
- 10.Matsuura E, Igarashi M, Igarashi Y, et al. Molecular definition of human β2-glycoprotein I (β2-GPI) by cDNA cloning and inter species differences of β2-GPI in alternation anticardiolipin binding. Int Immunol. 1991;3:1217–21. doi: 10.1093/intimm/3.12.1217. [DOI] [PubMed] [Google Scholar]
- 11.Hunt JE, Krilis S. The fifth domain of β2-glycoprotein I contains a phospholipid binding site (Cys281-Cys288) and a region recognized by anticardiolipin antibodies. J Immunol. 1994;152:653–9. [PubMed] [Google Scholar]
- 12.Igarashi M, Matsuura E, Igarashi Y, et al. Human β2-glycoprotein I as an anticardiolipin cofactor determined using deleted mutants expressed by a baculovirus system. Blood. 1996;87:3262–70. [PubMed] [Google Scholar]
- 13.Matsuura E, Igarashi Y, Yasuda T, et al. Anticardiolipin antibodies recognize β2-glycoprotein I structure altered by interacting with an oxygen modified solid phase surface. J Exp Med. 1994;179:457–62. doi: 10.1084/jem.179.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roubey RAS, Eisenberg RA, Harper MF, Winfield JB. ‘Anticardiolipin’ autoantibodies recognize β2-glycoprotein I in the absence of phospholipid. J Immunol. 1995;154:954–60. [PubMed] [Google Scholar]
- 15.Tsutsumi A, Matsuura E, Ichikawa K, et al. Antibodies to β2-glycoprotein I and clinical manifestations in patients with systemic lupus erythematosus. Arthritis Rheum. 1996;39:1466–74. doi: 10.1002/art.1780390905. [DOI] [PubMed] [Google Scholar]
- 16.Amengual O, Atsumi T, Khamashta M, et al. Specificity of ELISA for antibody to β2-glycoprotein I in patients with antiphospholipid syndrome. Br J Rheumatol. 1996;35:1239–43. doi: 10.1093/rheumatology/35.12.1239. [DOI] [PubMed] [Google Scholar]
- 17.Nimpf J, Wurm H, Kostner M. Interaction of β2-glycoprotein I with human blood platelets: influences upon the ADP induced aggregation. Thromb Haemost. 1985;54:397–401. [PubMed] [Google Scholar]
- 18.Schousboe I, Rasmussen MS. Synchronized inhibition of the phospholipid mediated autoactivation of Factor XII in plasma by β2-glycoprotein I and anti-β2-glycoprotein I. Thromb Haemostas. 1995;73:798–804. [PubMed] [Google Scholar]
- 19.Schousboe I. β2-glycoprotein I: plasma inhibitor of contact activation of the intrinsic blood coagulation pathway. Blood. 1985;66:1086–91. [PubMed] [Google Scholar]
- 20.Shi W, Chong B, Hogg P, Chesterman C. Anticardiolipin antibodies block the inhibition by β2-glycoprotein I of the factor Xa generating activity of platelets. Thromb Haemost. 1993;70:342–5. [PubMed] [Google Scholar]
- 21.Bancsi LF, van der Linden IK, Bertina RM. β2-glycoprotein I deficiency and the risk of thrombosis. Thromb Haemost. 1992;67:649–53. [PubMed] [Google Scholar]
- 22.Benedetti ED, Reber G, Miescher PA. No increase of β2-glycoprotein I levels in patients with antiphospholipid antibodies. Thromb Haemost. 1992;68:624. [PubMed] [Google Scholar]
- 23.Esmon CT. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J Biol Chem. 1989;264:4743–6. [PubMed] [Google Scholar]
- 24.Esmon CT, Owen WG. Identification of an endothelial cell cofactor for thrombin-catalysed activation of protein C. Proc Natl Acad Sci USA. 1981;78:2249–52. doi: 10.1073/pnas.78.4.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esmon NL, Owen WG, Esmon CT. Isolation of a membrane-bound cofactor for thrombin-catalyzed activation of protein C. J Biol Chem. 1982;257:859–64. [PubMed] [Google Scholar]
- 26.Suzuki K, Stenflo J, Dahlbäck B, Teodorsson B. Inactivation of human coagulation Factor V by activated protein C. J Biol Chem. 1983;258:1914–8. [PubMed] [Google Scholar]
- 27.Dahlbäck B. Protein S and C4b-binding protein: components involved in the regulation of the protein C anticoagulant system. Thromb Haemost. 1991;66:49–61. [PubMed] [Google Scholar]
- 28.Griffin JH, Evatt B, Zimmerman TS, et al. Deficiency of protein C in congenital thrombotic disease. J Clin Invest. 1981;68:1370–3. doi: 10.1172/JCI110385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertina RM, Koeleman BPC, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 30.Svensson PJ, Dahlbäck B. Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med. 1994;330:517–22. doi: 10.1056/NEJM199402243300801. [DOI] [PubMed] [Google Scholar]
- 31.de Groot PG, Derksen Rhwm. Protein C pathway, antiphospholipid antibodies and thrombosis. Lupus. 1994;3:229–33. doi: 10.1177/096120339400300405. [DOI] [PubMed] [Google Scholar]
- 32.Marciniak E, Romond EH. Impaired catalytic function of activated protein C: a new in vitro manifestation of lupus anticoagulant. Blood. 1989;71:2426–32. [PubMed] [Google Scholar]
- 33.Malia RG, Kitchen S, Greaves M, Preston FE. Inhibition of activated protein C and its cofactor protein S by antiphospholipid antibodies. Br J Haematol. 1990;76:101–7. doi: 10.1111/j.1365-2141.1990.tb07843.x. [DOI] [PubMed] [Google Scholar]
- 34.Borrell M, Sala N, de Castellarnau C, et al. Immunoglobulin fractions isolated from patients with antiphospholipid antibodies prevent the inactivation of factor Va by activated protein C on human endothelial cells. Thromb Haemost. 1992;68:101–7. [PubMed] [Google Scholar]
- 35.Oosting JD, Derksen Rhwm, Bobbink IWG, et al. Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S. An explanation for their pathogenic mechanism? Blood. 1993;81:2618–25. [PubMed] [Google Scholar]
- 36.Keeling DM, Wilson AJG, Mackie IJ, et al. Role of β2-glycoprotein I and anti-phospholipid antibodies in activation of protein C in vitro. J Clin Pathol. 1993;46:908–11. doi: 10.1136/jcp.46.10.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mori T, Takeya H, Nishioka J, et al. β2-glycoprotien I modulates the anticoagulant activity of activated protein C on the phospholipid surface. Thromb Haemost. 1996;75:49–55. [PubMed] [Google Scholar]
- 38.Simioni P, Lazzaro A, Zanadri S, Girolami A. Spurious protein C deficiency due to antiphospholipid antibodies. Am J Hematol. 1992;36:299–300. doi: 10.1002/ajh.2830360418. [DOI] [PubMed] [Google Scholar]
- 39.Matsuura E, Igarashi Y, Fujimoto M, et al. Heterogeneity of anticardiolipin antibodies defined by the anticardiolipin cofactor. J Immunol. 1992;148:3885–91. [PubMed] [Google Scholar]
- 40.Ichikawa K, Khamashta M, Koike T, et al. Reactivity of monoclonal anticardiolipin antibodies from patients with the antiphospholipid syndrome to β2-glycoprotein I. Arthritis Rheum. 1994;37:1453–61. doi: 10.1002/art.1780371008. [DOI] [PubMed] [Google Scholar]
- 41.Del PaPa N, Guidali L, Sala A, et al. Endothelial cells as target for antiphospholipid antibodies. Arthritis Rheum. 1997;40:551–61. doi: 10.1002/art.1780400322. [DOI] [PubMed] [Google Scholar]
- 42.Comp PC, de Bault LE, Esmon NL, Esmon CT. Human thrombomodulin is inhibited by IgG from two patients with non specific anticoagulants. Blood. 1983;62:299. (Abstr.). [Google Scholar]
- 43.Harris EN, Pierangeli SS. Functional effects of anticardiolipin antibodies. Lupus. 1996;5:372–7. doi: 10.1177/096120339600500507. [DOI] [PubMed] [Google Scholar]
- 44.Cariou R, Tobelem G, Soria C, Caen J. Inhibition of protein C activation by endothelial cells in the presence of lupus anticoagulant. N Engl J Med. 1986;314:1193–4. doi: 10.1056/NEJM198605013141817. [DOI] [PubMed] [Google Scholar]
- 45.Cariou R, Tobelem G, Bellucci S, et al. Effect of lupus anticoagulant on antithrombotic properties of endothelial cells—inhibition of thrombomodulin-dependent protein C activation. Thromb Haemost. 1988;60:54–58. [PubMed] [Google Scholar]
- 46.Taskiris DA, Settas L, Makris PE, Marbet GA. Lupus anticoagulant–antiphospholipid antibodies and thrombophilia. Relation to protein C–protein S–thrombomodulin. J Rheumatol. 1990;17:785–9. [PubMed] [Google Scholar]
- 47.Oosting JD, Derksen Rhwm, Hackeng TM, et al. In vitro studies of antiphospholipid antibodies and its cofactor β2-glycoprotein I, show negligible effects on endothelial cell mediated protein C activation. Thromb Haemost. 1991;66:666–71. [PubMed] [Google Scholar]
- 48.Forastiero R, Kordich L, Basilotta E, Carreras L. Differences in protein S and C4b-binding protein levels in different groups of patients with antiphosphoplipid antibodies. Blood Coag Fibrinol. 1994;5:609–16. [PubMed] [Google Scholar]
- 49.Ginsberg J, Demers C, Brill-Edwards P, et al. Acquired free protein S deficiency is associated with antiphospholipid antibodies and increased thrombin generation in patients with systemic lupus erythematosus. Am J Med. 1995;98:379–83. doi: 10.1016/S0002-9343(99)80317-9. [DOI] [PubMed] [Google Scholar]
- 50.Ames PRJ, Tommasino C, Iannaccone L, et al. Coagulation activation and fibrinolytic imbalance in subjects with idiopathic antiphospholipid antibodies—a crucial role for acquired free protein S deficiency. Thromb Haemost. 1996;76:190–4. [PubMed] [Google Scholar]
- 51.Mitchell CA, Rowell JA, Hau L, et al. A fatal thrombotic disorder associated with an acquired inhibitor of protein C. N Eng J Med. 1987;317:1620–42. doi: 10.1056/NEJM198712243172606. [DOI] [PubMed] [Google Scholar]
- 52.Ruiz-Argüelles A, Vazquez-Prado J, Deleze M, et al. Presence of serum antibodies to coagulation protein C in patients with systemic lupus erythematosus is not associated with antigenic or functional protein C deficiencies. Am J Hematol. 1993;44:58–59. doi: 10.1002/ajh.2830440112. [DOI] [PubMed] [Google Scholar]
- 53.Bakker HM, Tans G, Janssen-Claessen T, et al. The effect of phospholipids, calcium ions and protein S on rate constants of human factor Va inactivation by activated human protein C. Eur J Biochem. 1992;208:171–8. doi: 10.1111/j.1432-1033.1992.tb17171.x. [DOI] [PubMed] [Google Scholar]
- 54.Pei G, Powers DD, Lentz BR. Specific contribution of different phospholipid surfaces to the activation of prothrombin by the fully assembled prothrombinase. J Biol Chem. 1993;268:3226–33. [PubMed] [Google Scholar]
- 55.Smirnov MD, Esmon CT. Phosphatidylethanolamine incorporation into vesicles selectively enhances factor Va inactivation by activated protein C. J Biol Chem. 1994;269:816–9. [PubMed] [Google Scholar]
- 56.Simantov R, LaSala JM, Lo SK, et al. Activation of cultured vascular endothelial cells by antiphospholipid antibodies. J Clin Invest. 1995;96:2211–9. doi: 10.1172/JCI118276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tannenbaum SH, Finco R, Cines DB. Antibody and immune complexes induce tissue factor production by human endothelial cells. J Immunol. 1986;137:1532–7. [PubMed] [Google Scholar]
- 58.Atsumi T, Khamashta MA, Amengual O, Hughes GRV. Up-regulated tissue factor expression in antiphospholipid syndrome. Thromb Haemost. 1997;77:222–3. [PubMed] [Google Scholar]
- 59.Jurado M, Paramo JA, Gutierrez-Pimentel M, Rocha E. Fibrinolytic potential and antiphospholipid antibodies in systemic lupus erythematosus and other connective tissue disorders. Thromb Haemost. 1992;68:516–20. [PubMed] [Google Scholar]
- 60.Lellouche F, Martinuzzo M, Said P, et al. Imbalance of thromboxane/prostacyclin biosynthesis in patients with lupus anticoagulant. Blood. 1991;78:2894–9. [PubMed] [Google Scholar]
- 61.Arnout J. The pathogenesis of the antiphospholipid syndrome: a hypothesis based on parallelisms with heparin-induced thrombocytopenia. Thromb Haemost. 1996;75:536–41. [PubMed] [Google Scholar]
- 62.Smirnov MD, Triplett DT, Comp PC, et al. On the role of phosphatidylethanolamine in the inhibition of activated protein C activity by antiphospholipid antibodies. J Clin Invest. 1995;95:309–16. doi: 10.1172/JCI117657. [DOI] [PMC free article] [PubMed] [Google Scholar]