

Abstract
Glucocorticoids (GC) are potent anti-inflammatory and immunosuppressive agents that act on many cells of the body, including monocytes. Here we show that a 5-day course of high dose GC therapy differentially affected the CD14++ and the CD14+ CD16+ monocyte subpopulations in 10 patients treated for multiple sclerosis. While the classical (CD14++) monocytes exhibited a substantial increase from 495 ± 132 to 755 ± 337 cells/μl, the CD14+ CD16+ monocytes responded with a pronounced decrease from 36 ± 15 to 2 ± 3 cells/μl (P < 0.001). In 4/10 patients the CD14+ CD16+ monocytes fell below detection limits (< 0.2 cells/μl). This observation was confirmed when the CD14+ CD16+ monocytes were identified by virtue of their low CD33 expression as these cells decreased as well. After discontinuation of GC therapy the CD14+ CD16+ monocytes reappeared and reached normal levels after 1 week. The profound depletion of CD14+ CD16+ monocytes by GC as described here is a novel effect of GC action in vivo and may contribute to GC-mediated immunosuppression. Determination of the number of this monocyte subset may also serve to monitor the effectiveness of GC therapy in patients requiring immunosuppressive treatment.
Keywords: glucocorticoids, CD14+ CD16+ monocyte, immunosuppression, multiple sclerosis
INTRODUCTION
Glucocorticoids (GC) are physiological inhibitors of inflammatory responses and are widely used as immunosuppressive and anti-inflammatory agents. GC inhibit many steps in the inflammatory process, e.g. synthesis of cytokines such as tumour necrosis factor (TNF) [1,2], IL-1 [3,4] and IL-6 [5], expression of cell surface molecules required for immune functions [6,7] and migration of leucocytes into inflamed sites [8,9].
High dose GC therapy induces a short monocytopenic phase followed by transient monocytosis [10]. The monocytopenia is thought to be a consequence of reduced release of newly formed monocytes from the bone marrow [11,12], the following monocytosis can be explained by a diminished egress of these cells from the peripheral blood.
While these gross changes of monocyte numbers after GC therapy have been reported repeatedly, few studies are available regarding changes of monocyte subpopulations after GC administration [13,14].
In previous experiments we identified the subpopulation of CD14+ CD16+ monocytes in peripheral blood of man [15,16]. In contrast to the regular blood monocytes (CD14++), these cells are characterized by low expression of the endotoxin receptor (CD14) and additional expression of the Fcγ receptor type III (CD16). In healthy subjects the CD14+ CD16+ cells with 45 cells/μl blood account for about 10% of all monocytes. This subpopulation has been shown to represent a mature version of the regular monocytes [17]. Furthermore, it exhibits several features which may point towards an important role of this subset in generating a proinflammatory immune reaction, since these cells show a higher expression of HLA-DR, intercellular adhesion molecule-1 (ICAM-1) and they lack IL-10 synthesis [17,18]. Consistent with these observations, CD14+ CD16+ monocytes have been shown to be expanded in several inflammatory diseases such as sepsis [19], HIV infection [20,21] and tuberculosis [22].
Here, we report on the selective suppression of CD14+ CD16+ monocytes during anti-inflammatory therapy with high dose GC treatment.
PATIENTS AND METHODS
Patients
The study group consisted of 10 patients (two males, eight females, mean age 44 ± 11 years) who underwent high dose GC therapy (methylprednisolone (Urbason; Hoechst, Frankfurt, Germany) 500 mg intravenously for 5 days) for an acute exacerbation of multiple sclerosis (MS) at the Department of Neurology, Grosshadern Hospital, Munich University. No other immunosuppressive agent except GC was given before or during the observation period. Each patient gave informed consent. Therapy was started at 6 p.m. after the diagnosis had been established, and the following four doses were administered at 8 a.m. on the next four consecutive days. Blood leucocytes were analysed on day 0 (before therapy) and then at daily intervals whenever possible.
Three healthy volunteers (two males, one female) who had given informed consent were treated with a single dose of 250 mg methylprednisolone at 8 a.m. and cells were analysed by immunofluorescence before, 6, 12 and 24 h after medication.
Cytokine determination
Whole blood was rapidly spun at 4°C, 800 g for 5 min and plasma without anticoagulant was stored at −80°C. For cytokine analysis plasma was thawed at 37°C and coagulated by addition of glass beads. Serum (100 μl) was used for analysis of macrophage colony-stimulating factor (M-CSF) with a commercially available kit (M-CSF-ELISA; DPC Biermann, Bad Nauheim, Germany).
Leucocyte count
The absolute number of total leucocytes was analysed using a Coulter Counter T 840 (Coulter, Krefeld, Germany).
Immunofluorescence studies
Analysis of monocyte subpopulations was done essentially as detailed under http://www.med.uni-muenchen.de/imuno/ziegler. In brief, samples of EDTA-anticoagulated blood were drawn before initiation of GC therapy and at 8 a.m. before administration of the following GC doses. Samples were immediately stored on ice for a maximum of 2 h. Antibodies to the endotoxin receptor CD14 (My4-PE; Coulter), FcγRIII CD16 (3G8-Fitc; Coulter), HLA-DR (I2-Fitc; Coulter), CD33 (MD33.6-Fitc; CLB, Amsterdam, The Netherlands), CD11b (Bear 1-Fitc; Immunotech, Hamburg, Germany) or the respective isotypic controls were reacted under saturating conditions on ice with 100 μl of whole blood for 20 min. Erythrocytes were lysed and leucocytes fixed using the Coulter Q-Prep workstation. Cells were then washed once with cold PBS and immunofluorescence was analysed in a FACScan flow cytometer (Becton Dickinson, San Jose, CA). Leucocytes (13 000 events) or monocytes (5000 events) were acquired by gating on forward (FSC) and side angle scatter (SSC) properties. The percentage of non-viable cells detected by staining with propidium iodide (Sigma, Deisenhofen, Germany) was negligible (< 2%).
The number of total monocytes per μl blood was calculated as leucocytes/μl blood × percentage cells in monocyte scatter gate among all leucocytes × percentage of all CD14+ cells within the monocyte gate divided by 10 000. Number of CD14+ CD16+ monocytes/μl was calculated as total monocytes per μl blood × percentage CD14+ CD16+ monocytes among all CD14+ monocytes divided by 100.
Statistical analysis
Data obtained from the multiple individual donors are presented in tables and figures as the mean ± s.d. A paired Student's t-test was used. P < 0.05 was accepted as significant.
RESULTS
Kinetics of leucocytes and monocytes under GC therapy
In 5/10 donors initial leucocyte counts before GC administration were within normal limits, but five patients exhibited leucocytosis (> 10 000/μl) without any apparent infectious focus. On average, leucocytes of all donors (9880 ± 2255 cells/μl) were at the upper limit of healthy controls. GC treatment induced a marked leucocytosis, peaking on the second day (18 713 ± 4852 cells/μl; P ≤ 0.01) with subsequent normalization on day 9 (data not shown). At the end of GC therapy leucocytes were still clearly elevated (14 235 ± 3944 cells/μl).
Total monocyte count, i.e. CD14++ and CD14+ CD16+ cells (for definition see below), was 535 ± 139 cells/μl initially. Administration of GC was followed by transient monocytopenia on day 1 and then by monocytosis in 9/10 patients, with a mean increase in monocyte count by about factor 1.4 on days 4 and 5 (762 ± 341 cells/μl) (Fig. 1).
Fig. 1.
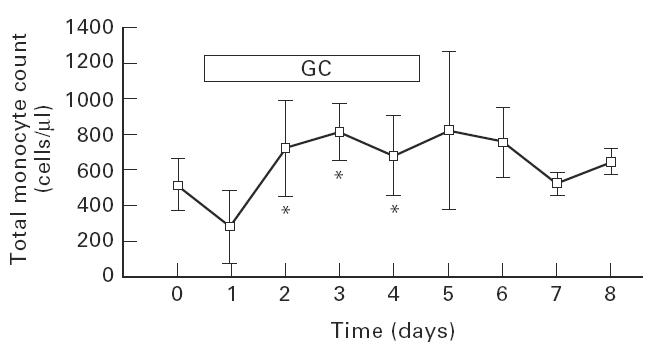
Effect of glucocorticoid therapy on total monocyte count in peripheral blood. Whole blood from patients before, during and up to 4 days after daily treatment with 500 mg methylprednisolone was stained with directly fluorochrome-conjugated monoclonal antibodies (PE–anti-CD14 and FITC–anti-CD16) and the absolute count of all monocytes, i.e. CD14++ plus CD14+ CD16+ monocytes, was determined by FACS. Given are mean ± s.d. for 10 patients with multiple sclerosis. The number of patients per time point varied from n = 10 to n = 3. *Significant compared with day 0 with P < 0.05.
Selective suppression of the CD14+ CD16+ monocytes by GC therapy
Next we analysed the effect of GC treatment on monocyte subpopulations that were defined by two-colour immunofluorescence using CD14 and CD16 antibodies.
Figure 2a depicts strongly CD14-positive regular monocytes and a population of double-positive cells that exhibit CD16 and low levels of CD14. After 5 days of GC therapy these CD14+ CD16+ cells were hardly detectable in peripheral blood (Fig. 2b). Figure 3 shows the time course for three representative examples (patients 1, 6 and 8) in which the CD14+ CD16+population was completely suppressed during GC therapy in peripheral blood. After discontinuation of GC these cells reappeared. On average, this population of monocytes, which accounted for 36 ± 15 cells/μl in all 10 patients before therapy, started to decrease already on day 1 after initiation of therapy. On day 4 or 5 the cells were strongly reduced to 2 ± 3 cells/μl (P < 0.001, Fig. 4). In 4/10 patients the CD14+ CD16+ monocytes fell below detection limits (< 0.2 cells/μl). This decrease was accompanied by a relative and absolute increase in regular CD14++ monocytes (495 ± 132 cells/μl versus 755 ± 337 cells/μl on day 4 or 5, NS).
Fig. 2.
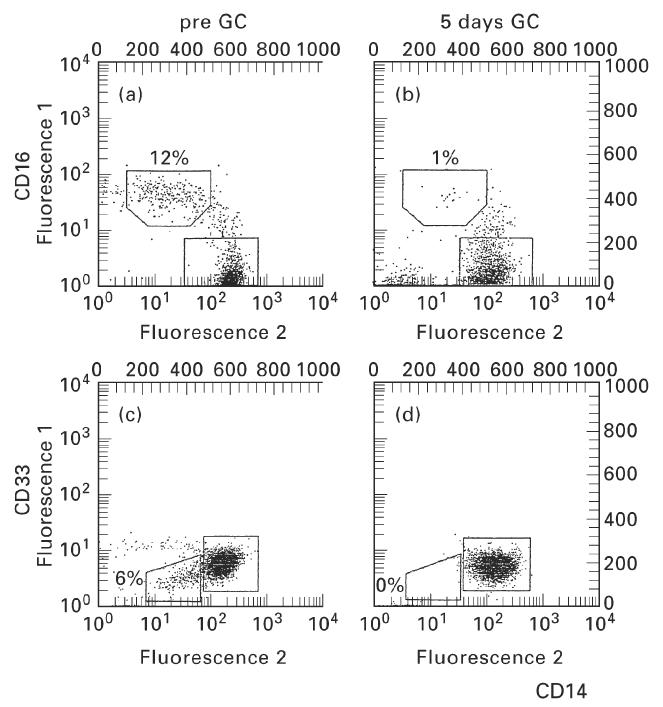
Depletion of CD14+ CD16+ monocytes by glucocorticoid (GC) therapy. Whole blood from patients before and after 5 days of daily treatment with 500 mg methylprednisolone was stained with directly fluorochrome-conjugated monoclonal antibodies (PE–anti-CD14 and FITC–anti-CD16 or FITC–anti-CD33) and 5000 cells per sample were analysed in the monocyte scatter gate by FACS. (a,b) Patient 3. (c,d) Patient 8. Absolute count of CD14+ CD16+ monocytes in patient 3 decreased from 52 cells/μl to eight cells/μl. CD14+ CD33+ monocytes in patient 8 decreased from 23 cells/μl to 0.3 cells/μl.
Fig. 3.
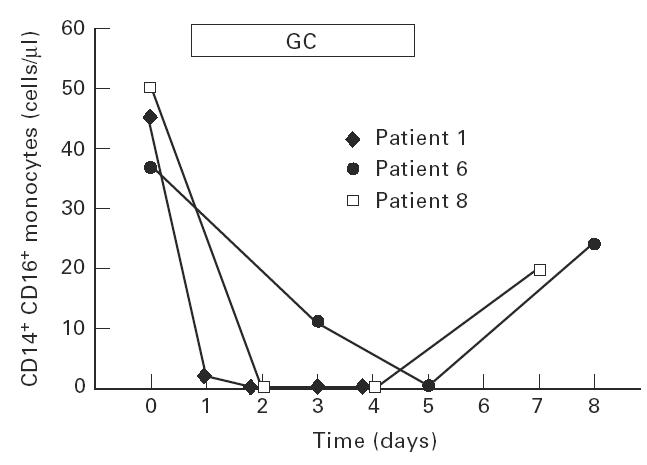
Time course of glucocorticoid (GC)-induced depletion of CD14+ CD16+ monocytes. The CD14+ CD16+ monocytes were determined by flow cytometry as given in the legend to Fig. 2.
Fig. 4.
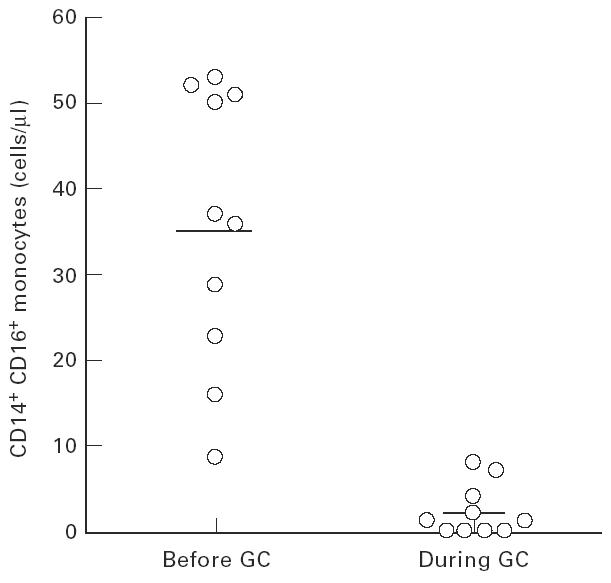
Effect of glucocorticoid (GC) therapy on CD14+ CD16+ monocytes. Cells were determined as given in the legend to Fig. 2. The average values ± s.d. were 36 ± 15 cells/μl before therapy and 2 ± 3 cells/μl on days 4 and 5. The difference is significant with P < 0.001.
The CD14+ CD16+ cells again increased in number after the end of therapy (Fig. 3) and reached control range approximately 1 week later (data not shown).
In addition to the 10 patients with MS, we studied one patient with glomerulonephritis and another with sarcoidosis who received a daily dose of 100 mg and 60 mg GC, respectively. Both patients responded with a depletion of CD14+ CD16+ monocytes from > 64 to < 9 cells/μl. Also, three apparently healthy volunteers who were treated with a single dose of 250 mg methylprednisolone showed a decrease of CD14+ CD16+ monocytes from 71 ± 18 cells/μl before treatment to 4 ± 2 cells/μl after 12 h. This indicates that depletion of CD14+ CD16+ monocytes is restricted neither to patients with MS nor to patients with inflammatory disease.
A significant spill-over of neutrophils which can also express CD14 and CD16 was excluded by carefully gating on the monocyte population in scatter properties and control staining with HLA-DR and CD33, both markers which are not expressed on neutrophils in blood (data not shown).
Effect of GC therapy on CD33+ monocytes
CD14+ CD16+ monocytes have previously been shown to express low levels of CD33 (CD33+) when compared with regular monocytes (CD33++) [17]. Therefore, CD14+ CD16+ cells can also be identified as CD14+ CD33+ cells. In order to demonstrate that GC treatment leads to a decrease of CD14+ CD16+ monocytes from peripheral blood and not just to a disappearance of the CD16 cell surface molecules on these cells, we analysed the kinetics of CD14+ CD33+ cells in peripheral blood. Again, we could demonstrate an almost complete suppression of these cells (Fig. 2, compare Fig. 2c and Fig. 2d). In the course of time these cells showed a pattern of decrease and increase similar to the CD14+ CD16+ cells. On average, the CD14+ CD33+ cells decreased from 26 ± 15 cells/μl on day 0 to 5 ± 4 cells/μl on day 4 or 5 (n = 8, P < 0.005).
Furthermore, Fig. 2c,d also demonstrates that the disappearance of the CD16+ and the CD33low cells does not lead to an increase of the CD14low cells that are CD16−and CD33−, respectively (data not shown). Also, the population of CD14−cells within the monocyte scatter remained constant (8 ± 4% before therapy and on days 4 and 5). Thus, the disappearance of the CD14+ CD16+ cells cannot simply be attributed to a loss of surface marker expression of these cells during GC therapy.
The CD16 antigen is also expressed on blood granulocytes. However, this expression was not affected by GC therapy (data not shown).
GC and surface molecule expression on regular monocytes
In order to assess other biological effects of GC on the expression of functionally important monocyte surface markers we studied the expression of HLA-DR, CD14, CD11b and CD33 on the regular CD14++ monocytes. There was no significant change in the membrane expression of CD11b, CD33 or CD14 during GC treatment (data not shown). Expression of CD14 by all CD14+ monocytes increased slightly due to the loss of the CD14low population (specific mean fluorescence intensity (sMFI) before 389 ± 57 versus 401 ± 53 on days 4 and 5).
However, GC induced a significant down-regulation of HLA-DR expression, reaching a nadir of specific mean fluorescence intensity on day 4 or 5 (pre-GC 277 ± 20 versus 135 ± 21 on day 4 or 5, n = 8, P < 0.001). After termination of GC treatment HLA-DR expression rapidly normalized within 1 week.
DISCUSSION
GCs are potent anti-inflammatory steroid hormones that act on a variety of cells in the body, e.g. endothelial cells and the different populations of leucocytes, including monocytes. GC act by binding to nuclear and probably also to cell surface receptors [23] and this is followed by reduced transcription of many proinflammatory genes [7,24,25]. This down-regulation of gene expression is probably mediated by the interference with NF-κB-mediated transactivation of these genes [26–29]. On the other hand, some genes are induced by GC and this includes macrophage migration inhibitory factor (MIF), IL-1 receptor type II (IL-1RII) and IL-10 [30–33].
MIF is a proinflammatory mediator which restores the cytokine production in LPS-stimulated macrophages treated with GC [31,34]. Therefore induction of this protein by GC is thought to counterbalance the immunosuppression brought about by GC. IL-1RII is considered a decoy receptor that binds IL-1 without signalling and thereby prevents this cytokine from binding to and activating through the IL-1RI [30]. IL-10 is a potent suppressive cytokine that down-regulates a whole series of proinflammatory genes [35]. Hence the induction by GC of these molecules also serves to suppress the immune response.
Other effects of GC that go in the same direction are the inhibition of cell adhesion and migration [6,8,9] and the induction of apoptosis in various types of leucocytes [36].
In the present study we report on an almost complete eradication of the subpopulation of CD14+ CD16+ monocytes in human peripheral blood. The CD14+ CD16+ monocytes [37] possess properties of tissue macrophages and share some features with alveolar macrophages [17]. Since these cells are effective producers of TNF but fail to synthesize detectable amounts of the anti-inflammatory cytokine IL-10 [18], the CD14+ CD16+ monocytes may be considered proinflammatory monocytes. Also these cells exhibit high levels of MHC class II and of adhesion molecules, all of which may facilitate T cell activation. The concept that the CD14+ CD16+ monocytes represent a proinflammatory type of cell is supported by the finding that CD14+ CD16+ monocytes are dramatically expanded in diseases with excessive inflammation like sepsis [19]. The disappearance of the CD14+ CD16+ monocytes after treatment with GC would therefore be in line with the anti-inflammatory action of these steroid hormones.
One might speculate that GC therapy merely down-regulates the surface expression of CD14 or CD16 on the CD14+ CD16+ monocytes, so that the cells become invisible when identified by virtue of their surface marker expression. In our patients we were unable to detect a significant change in CD14 expression on the CD14++ monocytes, and CD14 expression of all CD14+ monocytes even increased due to the loss of the CD14low population (data not shown). Furthermore, there was no increase of cells negative for CD14 when gating on monocytes according to scatter properties. Nockher et al. [38] recently reported on a reduced CD14 expression by peripheral blood monocytes in patients with acute inflammatory diseases receiving continuous GC therapy. In this study the effect of GC was, however, analysed beginning on day 7 of therapy. Hence, it is possible that the in vivo decrease of cell surface CD14 requires a prolonged period of treatment, so that it is not detectable within the first 5 days of therapy.
GC also do not lead to a general down-regulation of CD16, since expression of this cell surface molecule on granulocytes is unaffected in patients treated with methylprednisolone (data not shown). Also, in vitro experiments with cultured monocytes showed no decrease of surface CD16 during treatment with dexamethasone 10−6 m over a period of 5 days (data not shown). Furthermore, when CD14+ CD16+ monocytes were identified as low CD33 cells, then these cells were also depleted (Fig. 2). Taken together, these data indicate that GC therapy, in fact, leads to a depletion of the CD14+ CD16+ monocytes in peripheral blood.
As to the mechanism of this ablation, there are several possibilities. In vitro treatment with GC has been shown to increase adherence of RM3/1+ monocytes to endothelium [39]. Subsequent migration into tissues is, however, unlikely, since GC rather inhibit transmigration [9]. Apoptosis is an attractive possibility [36], but our studies using the TdT-mediated dUTP nick end labelling (TUNEL) technique failed to detect any evidence for this mechanism in peripheral blood monocytes of two patients and three healthy volunteers (data not shown).
With ongoing efflux of CD14+ CD16+ monocytes from the blood pool, treatment with GC may also operate by blocking the influx of new CD14+ CD16+ monocytes by blocking their differentiation.
Differentiation of leucocytes is governed by cytokines like the colony-stimulating factors, and M-CSF is an attractive candidate for induction of the CD14+ CD16+ monocytes, since patient treatment with M-CSF leads to a dramatic increase of these cells [40–44]. Levels of M-CSF in four patients, however, did not show any consistent pattern during GC therapy (data not shown). Still, we consider this kind of mechanism most likely and assume that other signals pertinent to monocyte/macrophage differentiation are blocked by GC.
The almost complete eradication of the CD14+ CD16+ monocytes is the most dramatic effect seen in peripheral blood. Most of this decrease occurs within the first 48 h of GC therapy. However, a more detailed analysis with hourly intervals early after initiation of GC therapy is required. The regular monocytes did, however, also respond to GC, since consistent with earlier reports [12] we observed an initial decrease followed by a pronounced increase in the number of these cells. Also, we detected a reduced expression of HLA-DR on the CD14++ monocytes. This is in line with in vitro data that show a GC-induced reduction in the expression of MHC class II molecules on monocytes [45]. Since DR molecules are central to antigen presentation and activation of T lymphocytes, this effect may contribute to the anti-inflammatory action of GC, as may the induction of the RM3/1 phenotype [13,14].
Depletion of CD14+ CD16+ monocytes is not restricted to patients with MS, since we observed a similar effect in one patient each with sarcoidosis and one with glomerulonephritis. Furthermore, in three healthy individuals GC treatment also resulted in depletion of these cells (data not shown), indicating that the effect of GC is not dependent on the presence of an inflammatory disease.
Doses of GC in the range of 60–100 mg methylprednisolone/day also seem to be effective in depleting the CD14+ CD16+ monocytes. However, a detailed study of the effects of different doses and of tapering of GC therapy is still required.
The CD14+ CD16+ monocytes were reported to be expanded in a variety of inflammatory conditions (reviewed in [37]), but thus far a decrease of these cells has only been observed in athletes after an ultramarathon run [46]. Here, the same mechanism as reported in the present study may operate, since excessive exercise is associated with a strong increase of endogenous GC [47].
When GC treatment was discontinued the CD14+ CD16+ monocytes returned to the initial level within 1 week. The reappearance of these cells may therefore be taken as an indication that the immune competence has recovered.
Several earlier reports have documented a dramatic expansion of CD14+ CD16+ monocytes in sepsis [19,48], which is a disease with excessive inflammation, while a more moderate increase of these cells was observed in tuberculosis [22], where inflammation is less pronounced. This may suggest that the number of CD14+ CD16+ monocytes in human peripheral blood may be used as an indicator of the severity of an inflammatory response.
On the other hand, when CD14+ CD16+ monocyte numbers decrease, as shown here with GC treatment, then this may be an indicator of the degree of immunosuppression, but the functional significance of this observation remains to be elucidated. Further studies are required to test the hypothesis that suppression of the CD14+ CD16+ subpopulation contributes to the immunosuppression by GC. For clinical purposes, the determination of the number of CD14+ CD16+ monocytes in blood may be useful for monitoring of the effectiveness of GC therapy.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (SFB 217) and by the VerUm Foundation. The authors acknowledge the expert technical assistance of A. Frank-Wanger and J. Köbler and the assistance in statistical analysis by B. Schmidt. We thank R. Hohlfeld (Department of Neurology, Klinikum Grosshadern, University of Munich) for provision of patient samples.
References
- 1.Beutler B, Krochin N, Milsark IW, Luedke C, Cerami A. Control of cachectin (tumor necrosis factor) synthesis: mechanisms of endotoxin resistance. Science. 1986;232:977–80. doi: 10.1126/science.3754653. [DOI] [PubMed] [Google Scholar]
- 2.Luedke CE, Cerami A. Interferon-γ overcomes glucocorticoid suppression of cachectin/tumor necrosis factor biosynthesis by murine macrophages. J Clin Invest. 1990;86:1234–40. doi: 10.1172/JCI114829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knudsen PJ, Dinarello CA, Strom TB. Glucocorticoids inhibit transcriptional and posttranscriptional expression of interleukin-1 in U937 cells. J Immunol. 1987;139:4129–34. [PubMed] [Google Scholar]
- 4.Lee SW, Tsou A-P, Chan H, Thomas J, Petrie K, Eugui EM, Allison AC. Glucocorticoids selectively inhibit the transcription of the interleukin 1β gene and decrease the stability of interleukin 1β mRNA. Proc Natl Acad Sci USA. 1988;85:1204–8. doi: 10.1073/pnas.85.4.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waage A, Slupphaug G, Shalaby R. Glucocorticoids inhibit the production of IL-6 from monocytes, endothelial cells and fibroblasts. Eur J Immunol. 1990;20:2439–43. doi: 10.1002/eji.1830201112. [DOI] [PubMed] [Google Scholar]
- 6.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of corticosteroids: the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1992;89:9991–5. doi: 10.1073/pnas.89.21.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van de Stolpe A, Caldenhoven E, Raaijmakers JAM, Van der Saag PT, Koenderman L. Glucocorticoid-mediated repression of intercellular adhesion molecule-1 expression in human monocytic and bronchial epithelial cell lines. Am J Respir Cell Mol Biol. 1993;8:340–7. doi: 10.1165/ajrcmb/8.3.340. [DOI] [PubMed] [Google Scholar]
- 8.Oda T, Katori M. Inhibition site of dexamethasone on extravasation of polymorphonuclear leukocytes in the hamster cheek pouch microcirculation. J Leukoc Biol. 1992;52:337–42. doi: 10.1002/jlb.52.3.337. [DOI] [PubMed] [Google Scholar]
- 9.Mancuso F, Flower RJ, Perretti M. Leukocyte transmigration, but not rolling or adhesion, is selectively inhibited by dexamethasone in the hamster post-capillary venule. Involvement of endogenous lipocortin 1. J Immunol. 1995;155:377–86. [PubMed] [Google Scholar]
- 10.Rinehart JJ, Sagone AL, Balcerzak SP, Ackerman GA, LoBuglio AF. Effects of corticosteroid therapy on human monocyte function. N Engl J Med. 1975;292:236–41. doi: 10.1056/NEJM197501302920504. [DOI] [PubMed] [Google Scholar]
- 11.Thompson J, van Furth R. The effect of glucocorticosteroids on the proliferation and kinetics of promonocytes and monocytes of the bone marrow. J Exp Med. 1973;137:10–21. doi: 10.1084/jem.137.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fauci AS, Dale DC. The effect of in vivo hydrocortisone on subpopulations of human lymphocytes. J Clin Invest. 1974;53:240–6. doi: 10.1172/JCI107544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zwadlo-Klarwasser G, Bent S, Haubeck HD, Sorg C, Schmutzler W. Glucocorticoid-induced appearance of the macrophage subtype RM3/1 in the peripheral blood of man. Int Arch Allergy Appl Immunol. 1990;91:175–80. doi: 10.1159/000235111. [DOI] [PubMed] [Google Scholar]
- 14.Zwadlo-Klarwasser G, Neubert R, Stahlmann R, Schmutzler W. Influence of dexamethasone on the RM 3/1-positive macrophages in the peripheral blood and tissues of a New World Monkey (The Marmoset Callithrix jacchus) Int Arch Allergy Immunol. 1992;97:178–80. doi: 10.1159/000236115. [DOI] [PubMed] [Google Scholar]
- 15.Ziegler-Heitbrock HWL, Passlick B, Flieger D. The monoclonal antimonocyte antibody My4 stains B lymphocytes and two distinct monocyte subsets in human peripheral blood. Hybridoma. 1988;7:521–7. doi: 10.1089/hyb.1988.7.521. [DOI] [PubMed] [Google Scholar]
- 16.Passlick B, Flieger D, Ziegler-Heitbrock HWL. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. [PubMed] [Google Scholar]
- 17.Ziegler-Heitbrock HWL, Fingerle G, Ströbel M, Schraut W, Stelter F, Schütt C, Passlick B, Pforte A. The novel subset of CD14+/CD16+ blood monocytes exhibits features of tissue macrophages. Eur J Immunol. 1993;23:2053–8. doi: 10.1002/eji.1830230902. [DOI] [PubMed] [Google Scholar]
- 18.Frankenberger M, Sternsdorf T, Pechumer H, Pforte A, Ziegler-Heitbrock HWL. Differential cytokine expression in human blood monocyte subpopulations: a polymerase chain reaction analysis. Blood. 1996;87:373–7. [PubMed] [Google Scholar]
- 19.Fingerle G, Pforte A, Passlick B, Blumenstein M, Ströbel M, Ziegler-Heitbrock HWL. The novel subset of CD14+/CD16+ blood monocytes is expanded in sepsis patients. Blood. 1993;82:3170–6. [PubMed] [Google Scholar]
- 20.Allen JB, Wong HL, Guyre PM, Simon GL, Wahl SM. Association of circulating receptor FcγRIII-positive monocytes in AIDS patients with elevated levels of transforming growth factor-β. J Clin Invest. 1991;87:1773–9. doi: 10.1172/JCI115196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Locher C, Vanham G, Kestens L, Kruger M, Ceuppens JL, Vingerhoets J, Gigase P. Expression patterns of Fcγ receptors, HLA-DR and selected adhesion molecules on monocytes from normal and HIV-infected individuals. Clin Exp Immunol. 1994;98:115–22. doi: 10.1111/j.1365-2249.1994.tb06616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanham G, Edmonds K, Qing L, et al. Generalized immune activation in pulmonary tuberculosis: co-activation with HIV infection. Clin Exp Immunol. 1996;103:30–34. doi: 10.1046/j.1365-2249.1996.907600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brann DW, Hendry LB, Mahesh VB. Emerging diversities in the mechanism of action of steroid hormones. J Steroid Biochem Mol Biol. 1995;52:113–33. doi: 10.1016/0960-0760(94)00160-n. [DOI] [PubMed] [Google Scholar]
- 24.Han J, Thompson P, Beutler B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J Exp Med. 1990;172:391–4. doi: 10.1084/jem.172.1.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Standiford TJ, Kunkel SL, Rolfe MW, Evanoff HL, Allen RM, Strieter RM. Regulation of human alveolar macrophage- and blood monocyte-derived interleukin-8 by prostaglandin E2 and dexamethasone. J Respir Cell Mol Biol. 1992;6:75–81. doi: 10.1165/ajrcmb/6.1.75. [DOI] [PubMed] [Google Scholar]
- 26.Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NFκB activity through induction of IκB synthesis. Science. 1995;270:286–90. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 27.Scheinman RI, Cogswell PC, Lofquist AK, Baldwin ASJ. Role of transcriptional activation of IκBα in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–6. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 28.Adcock IM, Brown CR, Gelder CM, Shirasaki H, Peters MJ, Barnes PJ. Effects of glucocorticoids on transcription factor activation in human peripheral blood mononuclear cells. Am J Physiol. 1995;268:C331–8. doi: 10.1152/ajpcell.1995.268.2.C331. [DOI] [PubMed] [Google Scholar]
- 29.Brostjan C, Anrather J, Csizmadia V, Stroka D, Soares M, Bach FH, Winkler H. Glucocorticoid-mediated repression of NF-κB activity in endothelial cells does not involve induction of IκBα synthesis. J Biol Chem. 1996;271:19612–6. doi: 10.1074/jbc.271.32.19612. [DOI] [PubMed] [Google Scholar]
- 30.Re F, Muzio M, De Rossi M, Polentarutti N, Giri JG, Mantovani A, Colotta F. The type II ‘receptor’ as a decoy target for interleukin 1 in polymorphonuclear leukocytes: characterization of induction by dexamethasone and ligand binding properties of the released decoy receptor. J Exp Med. 1994;179:739–43. doi: 10.1084/jem.179.2.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 32.Tabardel Y, Duchateau J, Schmartz D, Marecaux G, Shahla M, Barvais L, Leclerc JL, Vincent JL. Corticosteroids increase blood interleukin-10 levels during cardiopulmonary bypass in men. Surgery. 1996;119:76–80. doi: 10.1016/s0039-6060(96)80217-0. [DOI] [PubMed] [Google Scholar]
- 33.Marchant A, Amraoui Z, Gueydan C, et al. Methylprednisolone differentially regulates IL-10 and tumour necrosis factor (TNF) production during murine endotoxaemia. Clin Exp Immunol. 1996;106:91–96. doi: 10.1046/j.1365-2249.1996.d01-799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernhagen J, Calandra T, Mitchell RA, et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–9. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 35.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, deVries JE. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–20. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montague JW, Cidlowski JA. Glucocorticoid-induced death of immune cells: mechanisms of action. Curr Top Microbiol Immunol. 1995;200:51–65. doi: 10.1007/978-3-642-79437-7_4. [DOI] [PubMed] [Google Scholar]
- 37.Ziegler-Heitbrock HWL. Heterogeneity of human blood monocytes: the CD14+CD16+ subpopulation. Immunol Today. 1996;17:424–8. doi: 10.1016/0167-5699(96)10029-3. [DOI] [PubMed] [Google Scholar]
- 38.Nockher WA, Scherberich JE. Expression and release of the monocyte lipopolysaccharide receptor antigen CD14 are suppressed by glucocorticoids in vivo and in vitro. J Immunol. 1997;158:1345–52. [PubMed] [Google Scholar]
- 39.Wenzel I, Roth J, Sorg C. Identification of a novel surface molecule, RM3/1, that contributes to the adhesion of glucocorticoid-induced human monocytes to endothelial cells. Eur J Immunol. 1996;26:2758–63. doi: 10.1002/eji.1830261131. [DOI] [PubMed] [Google Scholar]
- 40.Munn DH, Garnick MB, Cheung N-KV. Effects of parenteral recombinant human macrophage colony-stimulating factor on monocyte number, phenotype, and antitumor cytotoxicity in nonhuman primates. Blood. 1990;75:2042–8. [PubMed] [Google Scholar]
- 41.Weiner LM, Li W, Holmes M, Catalano RB, Dovnarsky M, Padavic K, Alpaugh RK. Phase I trial of recombinant macrophage colony-stimulating factor and recombinant γ-interferon: toxicity, monocytosis, and clinical effects. Cancer Res. 1994;54:4084–90. [PubMed] [Google Scholar]
- 42.Saleh MN, Goldman SJ, LoBuglio AF, et al. CD16+ monocytes in patients with cancer: spontaneous elevation and pharmacologic induction by recombinant human macrophage colony-stimulating factor. Blood. 1995;85:2910–7. [PubMed] [Google Scholar]
- 43.Schmid I, Baldwin GC, Jacobs EL, Isacescu V, Neagos N, Giorgi JV, Glaspy JA. Alterations in phenotype and cell-surface antigen expression levels of human monocytes: differential response to in vivo administration of rhM-CSF or rhGM-CSF. Cytometry. 1995;22:103–10. doi: 10.1002/cyto.990220205. [DOI] [PubMed] [Google Scholar]
- 44.Munn DH, Bree AG, Beall AC, et al. Recombinant human macrophage colony-stimulating factor in non-human primates: selective expansion of a CD16+ monocyte subset with phenotypic similarity to primate natural killer cells. Blood. 1996;88:1215–24. [PubMed] [Google Scholar]
- 45.Hawrylowicz CM, Guida L, Paleolog E. Dexamethasone up-regulates granulocyte-macrophage colony-stimulating factor receptor expression on human monocytes. Immunology. 1994;83:274–80. [PMC free article] [PubMed] [Google Scholar]
- 46.Gabriel H, Urhausen A, Brechtel L, Müller H-J, Kindermann W. Alterations of regular and mature monocytes are distinct, and dependent of intensity and duration of exercise. Eur J Appl Physiol. 1994;69:179–81. doi: 10.1007/BF00609414. [DOI] [PubMed] [Google Scholar]
- 47.Nieman DC, Berk LS, Simpson-Westerberg M, Arabatzis K, Youngberg S, Tan SA, Lee JW, Eby WC. Effect of long-endurance running on immune system parameters and lymphocyte function in experienced marathoners. Int J Sports Med. 1989;10:317–23. doi: 10.1055/s-2007-1024921. [DOI] [PubMed] [Google Scholar]
- 48.Blumenstein M, Boekstegers P, Fraunberger P, Andreesen R, Ziegler-Heitbrock HWL, Fingerle-Rowson G. Cytokine production precedes the expansion of CD14+CD16+ monocytes in human sepsis: a case report of a patient with self-induced septicemia. Shock. 1997;8:73–75. doi: 10.1097/00024382-199707000-00012. [DOI] [PubMed] [Google Scholar]