

Abstract
A unique cytokine, human cytotoxic factor (hCF), has been shown to occur in the sera of patients with dengue fever (DF) and dengue haemorrhagic fever (DHF). The present study was undertaken to investigate the ability of fresh PBMC of such patients to produce hCF. The PBMC were cultured for 24 h and the culture supernatants (CS) were analysed for the presence of hCF by cytotoxicity assay, competitive ELISA and dot blot tests. In 90% of 246 cases CS were positive for hCF by the three tests. CS were positive for hCF in PBMC collected from days 1–20 of illness but not at later periods. Higher cytotoxic activity was observed in CS of days 1–4 of illness and was highest in cases of DHF grade IV and lowest in cases of DF. Dot blot hybridization of RNA extracted from the PBMC of the patients showed the presence of mRNA for hCF in 94% of cases. A similar number of patients showed the presence of hCF in situ in the PBMC smears by fluorescent antibody technique. hCF was found only in CD4+ T cells. The findings thus present direct evidence of the production of hCF by CD4 T cells of cases of DF/DHF.
Keywords: dengue haemorrhagic fever, dengue virus, cytotoxic factor, cytokine
INTRODUCTION
The cytotoxic factor (CF) is a unique cytokine that is produced both in mice (mCF) and man (hCF) during dengue virus infection [1,2]. The mCF is similar to hCF in most of its properties, including molecular mass, antigenicity and reaction of their mRNA to the same oligonucleotide probe in Northern blot tests [3]. The amino-terminal sequence of mCF does not match with any other known protein or cytokine sequence, including IL-17 (database check end 1996). mCF and hCF appear to be pathogenesis-related proteins, capable of reproducing the pathological changes that are seen in cases of dengue haemorrhagic fever (DHF), e.g. increased capillary permeability, cerebral oedema and blood leucocyte changes [2,4–7].
The presence of hCF was first shown in the sera of 12 out of 19 cases of DHF in an epidemic at Shahjahanpur (UP, India) during 1993 [8]. The findings were later validated on 220 sera of such cases stored at AFRIMS, Bangkok, by purification and characterization of hCF [2].This was further confirmed by production of hCF after experimental in vitro infection of human PBMC with dengue virus [9]. An extensive epidemic of DHF occurred in Northern India, including Delhi and Lucknow, during August–November 1996. This provided an opportunity to extend further the studies on hCF. Production of a cytokine should be demonstrated by different approaches to overcome the drawbacks of various methods [10]. The present study describes ex vivo production of hCF by CD4+ T cells of PBMC of DF/DHF cases by a variety of methods.
PATIENTS AND METHODS
Cases
Patients suffering from typical dengue-like illness admitted to the Gandhi Memorial and Associated Hospitals, Lucknow, and the Paediatrics Department of the All India Institute of Medical Sciences, New Delhi, were studied. They were classified into those of dengue fever (DF) or DHF of grades I, II, III or IV according to the World Health Organization criteria [11]. A total of 246 patients were included in the study. The diagnosis of dengue virus infection was established either by virus isolation or by detection of virus-specific IgM in serum. From four cases, two from Lucknow and two from Delhi, dengue type 2 virus was isolated, while in the remaining cases detection of specific IgM was the basis of diagnosis (unpublished data). For controls, 50 age-matched healthy individuals, without history of any febrile or other illnesses in the previous 3 months, were included.
Preparation of PBMC
Peripheral venous blood was collected in a heparinized tube from cases of various grades of illness and normal healthy controls. The leucocyte-rich plasma was removed and the mononuclear cells (PBMC) consisting of monocytes and lymphocytes were separated on Lymphoprep, density 1.077 g/ml (Nyegaard & Co. As., Oslo, Norway). The cells collected from the interface layer were washed three times with Hanks' buffered salt solution (HBSS) and counted as described earlier [10].
The cells were used for culture, extraction of RNA, and smear preparation for Giemsa staining and fluorescent antibody test. In a number of cases smears were also prepared directly from the blood.
Preparation of enriched cell subpopulations
The PBMC were passed through a glass wool column to remove monocytes and dead cells and then filtered through a nylon wool column to prepare enriched T and B cell subpopulations [12]; their purity was checked as described [9,13]. To prepare monocyte-enriched suspensions, the PBMC were suspended in Eagle's minimum essential medium (MEM) containing 5% fetal calf serum (FCS) and layered in 4-cm Petri dishes (Nunclon, Nunc, Roskilde, Denmark) and incubated at 37°C in presence of 5% CO2/air. After 2 h the adherent cells were separated and their purity tested [13]. T cells were further enriched into CD4+ or CD8+ cells by treatment with mouse anti-human CD4 or CD8 MoAbs (Dako A/S, Glostrup, Denmark) and rabbit complement as described [9]. The cells obtained by different procedures described above were then washed and counted.
Preparation of cell cultures
PBMC (1 × 106 cells/ml) were cultured in Eagle's MEM containing 5% FCS using 4-cm Petri dishes (Nunclon, Nunc). The cultures were incubated at 37°C in the presence of 5% CO2/air. All cultures were set up in triplicate and were harvested after 24 h incubation. The culture supernatants were separated from cells by centrifugation at 3000 g at 4°C for 10 min and stored at −70°C until testing. The culture supernatants were screened for the presence of hCF by cytotoxicity, ELISA and dot blot tests. On a number of samples Western blot tests were also done [2,9].
Assay of cytotoxic activity
Culture supernatants collected from the cells obtained from cases of DF/DHF (CS) or normal healthy controls (NCS) were screened for the presence of hCF by the cytotoxicity assay as described elsewhere [1,5]. Briefly, equal volumes of CS/NCS and single-cell suspension of normal mouse spleen (2 × 106 cells) were mixed in 96-well perspex plate and kept at 4°C for 1 h. Viability of the cells was screened using trypan blue dye, and the percentage of non-viable cells was expressed after deduction of background values obtained from untreated control cells. The mean value + 2 s.d. obtained with NCS of 50 normal healthy controls was considered as ‘cut-off’ value and was 10%.
ELISA
This test was carried out as described elsewhere [8] using 1:5000 dilution of hCF-specific antibody [2] on the solid phase. The anti-hCF antibody used neutralized the cytotoxic activity of hCF in vitro, inhibited hCF-induced increase in capillary permeability in mice (up to 1:10 000 dilution) and reacted (up to 1:5000 dilution) in the Western blot tests and ELISA [2,3]. The controls included were purified hCF as positive control and a heterologous protein as negative control. The optical density (OD) values from wells coated with antibody without addition of any antigen were considered as 100% binding of the Protein A + horseradish peroxidase (HRP), while the wells without antibody coating yielded blank values. hCF present in the test sample bound with the antibody on the solid phase and inhibited subsequent binding of the Protein A + HRP, thus indicating presence of hCF. The percentage inhibition was calculated as follows: % inhibition = 100 − (OD with test sample − OD of blank)/(OD without test sample −OD of blank) × 100 [8]. The mean value + 2 s.d. obtained from the 50 normal healthy controls was considered as ‘cut-off’ value and was 25%.
Immunoblotting
The technique described earlier [2,9] was used. CS were blotted on nitrocellulose membrane and blocked. The blots were treated with anti-hCF antibody followed by treatment with anti-mouse IgG + HRP. For controls the blots were treated with anti-dengue virus antibody or normal mouse sera in place of anti-hCF antisera. The blots were developed using diaminobenzidine (Sigma Chemical Co., St Louis, MO) and hydrogen peroxide.
Extraction of RNA and Northern blot analysis
Total RNA was isolated from the cells by the acid guanidinium-thiocyanate-phenol chloroform method of Chomczynski & Sacchi [14]. RNA was quantified spectrophotometrically and its integrity examined by electrophoresis. The Northern blot analysis was done using 32P-labelled probe as described earlier [9], and autoradiograms were prepared. The two oligonucleotides used were: 5′ TGG CTC TAG ACA 3′ and 5′ TGG TTC TAG ACA 3′ and were prepared by the CSIR Centre for Biotechnology (New Delhi, India) [9].
Fluorescent antibody technique
The technique of Sander et al. [10] was used for intracellular localization of hCF. Briefly, the smears prepared from the PBMC of DF/DHF cases and healthy controls were fixed in 4% paraformaldehyde and 0.1% saponin and treated with hCF-specific antibody and prepared for indirect fluorescent study [15]. For controls irrelevant antisera and normal mouse sera were used. The smears were examined in transmitted light using a Leitz Dialux 20 fluorescent microscope.
RESULTS
Presence of hCF in PBMC
The supernatants (CS) from PBMC cultures of 246 patients were screened for the presence of hCF by ELISA, dot blot and cytotoxicity assay. A representative dot blot reaction is presented in Fig. 1. The findings presented in Table 1 show that all the CS from the cases of DHF grade III and DHF grade IV were positive for hCF, while the positivity was 91–95% in cases of DF and DHF grades I and II. Figure 2 demonstrates the presence of hCF from day 1 of onset of the fever up to day 20 of illness. None of the five CS of the cells collected during days 21–26 of illness were positive. Higher cytotoxic activity was observed during the first 4 days of illness. An analysis of the data of the extent of the cytotoxicity in relation to the grade of illness is presented in Fig. 3. It was observed that the mean cytotoxic activity in the NCS obtained from PBMC of normal healthy controls was 6 ± 2%, while it was 15 ± 3% in cases of DF and 30 ± 5% in DHF IV (P < 0.001).
Fig. 1.

A typical dot blot test for detection of human cytotoxic factor (hCF) in the culture supernatants (CS) of PBMC of the cases of various grades of dengue illness and normal healthy controls. Cultures were prepared (1 × 106 cells/ml) in 4-cm Nunc Petri dishes and incubated at 37°C in the presence of 5% CO2 and harvested after 24 h. CS were blotted on nitrocellulose membrane and were blocked. Blots were treated with anti-hCF antibody followed by anti-mouse IgG + horseradish peroxidase and then developed with substrate diaminobenzidine as described in Patients and Methods. The controls included CS from normal healthy individuals' PBMC (lane 9, A–H; lane 11, F–H); and purified hCF (1 μg/ml and its doubling dilutions) as positive control (lane 10, A–D).
Table 1.
Presence of human cytotoxic factor (hCF) in the culture supernatants of the PBMC of the cases of various grades of dengue illness
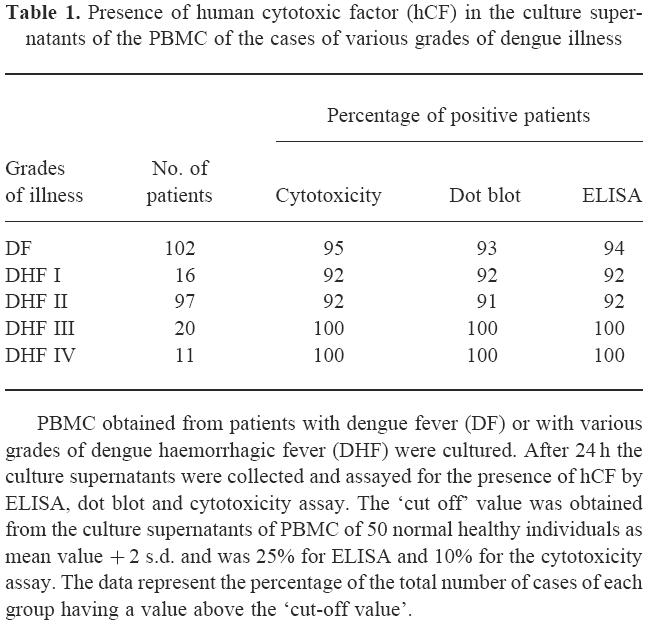
Fig. 2.

Presence of human cytotoxic factor (hCF) in the culture supernatants (CS) of PBMC collected on different days after the onset of illness. ▪, ELISA; □, dot blot; ♦, cytotoxicity assay. The ‘cut off’ value was obtained from the culture supernatants of PBMC of 50 normal healthy individuals as mean value + 2 s.d. and was 25% for ELISA and 10% for the cytotoxicity assay. The figures in parentheses represent the total number of patients in each group.
Fig. 3.
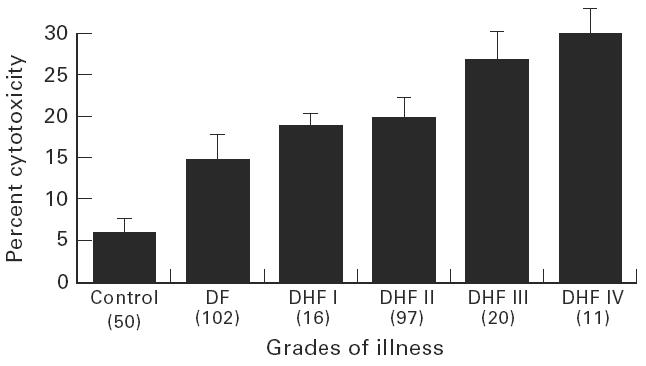
Mean cytotoxic activity in the culture supernatants from controls and cases of various grades of illness. The bars over the column represent the size of the s.d. and the figures in parentheses represent total number of patients in each group.
Presence of mRNA for hCF in PBMC
The findings presented in Fig. 4 show a typical autoradiogram of the dot blot hybridization analysis of mRNA for hCF. PBMC from cases of DF/DHF gave a positive reaction with hCF-specific oligonucleotide probe. hCF-specific mRNA was present in 94% of the cases. The normal healthy control PBMC gave a faint reaction.
Fig. 4.
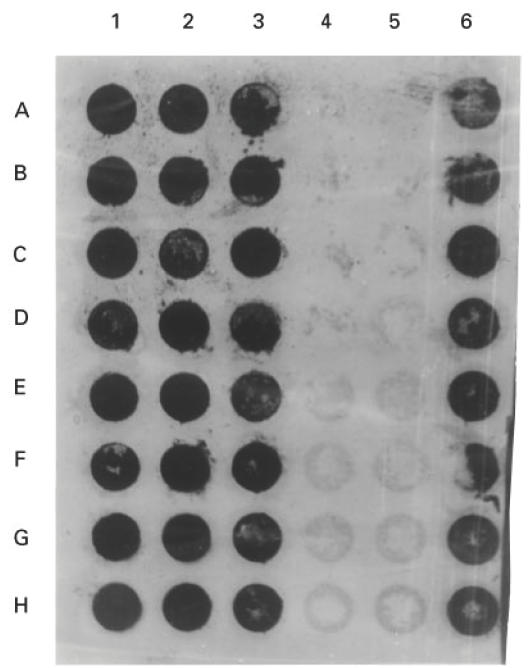
Dot blot hybridization analysis of total RNA isolated from PBMC of the cases of dengue fever (DF) (lane 1, A–D), DHF I (lane 1, E–H), DHF II (lane 2, A–D), DHF III (lane 2, E–H), DHF IV (lane 3, A–H), normal healthy controls (lane 4, A–H; lane 5, A–H), and dengue type 2 virus-infected mouse spleens (lane 6, A–H) as positive control. Each spot represents 5 μg of RNA from each patient/control or from mouse spleen blotted on nitrocellulose membrane. Hybridization was done as described in Patients and Methods.
Intracellular localization of hCF in PBMC
Smears prepared from PBMC were screened for the presence of hCF by indirect fluorescent antibody test. A typical positive cell showed localized clumpy cytoplasmic collections and also some staining of the cell membrane (data not shown). Presence of the hCF-positive cells was seen in the PBMC from day 1 of the illness to day 20, but not at later periods. The count was four to six positive cells/1000 cells at different time periods and in the cases of various grades of the illness. The hCF-positive cells were found in 93% of the DF/DHF cases, while cells from all normal healthy controls were negative. A false-positive reaction may be given by a cell that non-specifically adsorbs hCF from the environment. Therefore, to distinguish the hCF-producing cell from that with hCF adsorbed, a pilot experiment was done where PBMC from normal healthy controls were incubated with purified hCF for 1 h followed by screening by fluorescent antibody technique. These cells showed a weak diffuse staining, mainly of the cell membrane. This control confirmed the presence of hCF-producing cells in the PBMC of the cases of DF/DHF. Further, study of the enriched subpopulations of the cells showed the presence of hCF only in the CD4+ T cells and not in the CD8+ T cells, B cells or monocytes (Table 2).
Table 2.
Presence of human cytotoxic factor (hCF) in the PBMC of the cases of various grades of dengue illness as shown by the indirect fluorescent antibody test
DISCUSSION
The significant finding of the present study is the direct demonstration of the production of hCF by PBMC of cases of DF and DHF. It was observed that fresh PBMC obtained from cases continued to secrete hCF in vitro without any stimulation. The presence of hCF in the culture supernatants (CS) was detected by ELISA and cytotoxicity tests, and was confirmed by the dot blot tests in all cases, and by Western blot in a number of cases by the technique described earlier [2]. Further confirmation was obtained by (i) in situ demonstration of hCF in PBMC by fluorescent antibody test, and (ii) hCF-specific mRNA by Northern hybridization.
Another important finding of the present study is the presence of hCF in the CS and the cells from day 1 of illness to day 20, with peak amounts of hCF during the first 4 days, the mean cytotoxic activity being 25–30%. This is supported by our findings in the mouse model. Inoculation of mice with dengue virus intraperitoneally results in production of mCF, which is detectable on days 5–6 post-infection (p.i), reaches a peak on days 9–11 and then gradually declines and is not detectable after day 16 p.i [7]. Similarity with the mouse pattern is clear if we take into consideration the incubation period of dengue virus in man. So far the indications are that hCF is an indicator protein of dengue virus infection, as its production could not be induced in mice by inoculation of different mitogens or viruses, including Japanese encephalitis or Coxsackie B viruses, etc. Further presence of hCF could not be demonstrated in patients' sera of various bacterial or viral infections, including patients with typhoid fever, Japanese encephalitis, etc. ([2,3] and U. C. Chaturvedi, unpublished data). If this is true, hCF could become a useful diagnostic tool due to its presence in 90–100% of cases from day 1 to day 20.
Expression of mRNA is a much more sensitive and specific technique for the detection of a cytokine [16]. This technique has been successfully used for demonstration of the production of hCF by PBMC infected in vitro with dengue virus [9]. The findings of the present study, while supporting the above study, show the presence of hCF-specific mRNA in 94% of the cases of DF/DHF. In another study on cases of DF/DHF we have shown the production of hCF by, and presence of hCF-specific mRNA in, CD4+ T cells and not in CD8+ T cells, B cells or monocytes [17]. This confirmed the production of hCF by CD4+ T cells in the PBMC of the cases of DF/DHF. Another good method, the intracellular measurement of hCF, was tried using indirect fluorescent antibody technique. This technique has been used for various cytokines in PBMC [18]. The findings described here show the presence of hCF-positive cells in the PBMC of the cases of DF/DHF and not in those of normal healthy controls. Further, more intracellular localization of hCF was seen only in CD4+ T cells. No significant difference was found among the cases of various grades of the illness, neither by assay for the mRNA nor by intracellular localization of hCF. These are highly sensitive techniques to show a cytokine-producing cell. The presence of cytokine mRNA does not necessarily mean that the RNA is being translated or indicate the amount of cytokine secreted.
The pathogenic role of mCF and hCF has been established in producing lesions in mice that are seen in cases of DHF, e.g. increased capillary permeability, cerebral oedema and alterations in the number and functions of leucocytes [2,4–7]. Certain findings of the present study strengthened this view, for example: (i) a higher cytotoxic activity in cases of DHF grade IV than that of DF (P < 0.001); and (ii) higher cytotoxic activity up to day 4, coinciding with the onset of clinical illness. This indicates the need for quantification of hCF in various grades of the illness. The number of the hCF-producing cells may not indicate the amount of hCF produced. This will depend upon the factors, if any (not yet known), triggering them to secrete more hCF.
The findings of the present study confirm and extend our finding of the production of hCF by CD4+ T cells of PBMC during dengue virus infection in man, and have strengthened the evidence of its pathogenic role. Cloning of the gene for CF is urgently needed.
Acknowledgments
We are grateful to Drs A. K. Tripathi and K. L. Srivastava of K. G. Medical College, Lucknow, and Professor M. K. Bhan of the All India Institute of Medical Sciences, New Delhi, for permission to study their cases. The study was carried out with the financial assistance of the Indian Council of Medical Research, New Delhi.
References
- 1.Chaturvedi UC, Bhargava A, Mathur A. Production of cytotoxic factor in the spleen of dengue virus infected mice. Immunol. 1980;40:665–71. [PMC free article] [PubMed] [Google Scholar]
- 2.Mukerjee R, Chaturvedi UC, Vaughn DW, Kalayanarooj S. Purification and pathogenicity of the cytotoxic factor from the cases of dengue haemorrhagic fever. Curr Sci. 1997;72:494–501. [Google Scholar]
- 3.Mukerjee R, Chaturvedi UC. Cytokine antagonism by active vaccination. Cur Sci. 1995;69:900–2. [Google Scholar]
- 4.Khanna M, Chaturvedi UC, Sharma MC, Pandey VC, Mathur A. Increased capillary permeability mediated by a dengue virus-induced lymphokine. Immunol. 1990;69:449–53. [PMC free article] [PubMed] [Google Scholar]
- 5.Khanna M, Chaturvedi UC. Purification and amino-terminal sequence of the dengue virus-induced cytotoxic factor. Int J Exp Pathol. 1992;73:43–49. [PMC free article] [PubMed] [Google Scholar]
- 6.Chaturvedi UC, Gulati L, Mathur A. Inhibition of E-rosettes formation and phagocytosis by human blood leucocytes after treatment with dengue virus-induced cytotoxic factor. Immunol. 1982;45:679–84. [PMC free article] [PubMed] [Google Scholar]
- 7.Chaturvedi UC, Dhawan R, Khanna M, Mathur A. Breakdown of the blood–brain barrier during dengue virus infection of mice. J Gen Virol. 1991;72:859–66. doi: 10.1099/0022-1317-72-4-859. [DOI] [PubMed] [Google Scholar]
- 8.Mukerjee R, Chaturvedi UC. ELISA for detection of dengue virus-induced cytokine and its antibody. Ind J Exp Biol. 1997;35:225–31. [PubMed] [Google Scholar]
- 9.Mukerjee R, Chaturvedi UC, Dhawan R. Dengue virus- induced cytotoxic factor: production by peripheral blood leucocytes in vitro. Clin Exp Immunol. 1995;102:262–7. doi: 10.1111/j.1365-2249.1995.tb03775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sander B, Andersson J, Andersson U. Assessment of cytokines by immunofluorescence and the paraformaldehyde-saponin procedure. Immunol Rev. 1991;119:65–93. doi: 10.1111/j.1600-065x.1991.tb00578.x. [DOI] [PubMed] [Google Scholar]
- 11.Nimmannitya S. Clinical manifestations of dengue/dengue haemorrhagic fever. In: Thongcharoen P, editor. Dengue/dengue haemorrhagic fever. New Delhi: WHO-SEARO; 1993. pp. 48–54. [Google Scholar]
- 12.Trizio D, Cudkowicz G. Separation of T and B lymphocytes by nylon wool columns: evaluation of efficacy by functional assays in vivo. J Immunol. 1974;113:1093–7. [PubMed] [Google Scholar]
- 13.Khare M, Chaturvedi UC. Transmission of dengue virus-specific suppressor signal depends on the presence of calcium. Indian J Med Res. 1995;102:1–8. [PubMed] [Google Scholar]
- 14.Chomczynski P, Sacchi N. Single step method of RNA isolation by acid guanidinium thiocyanate phenol chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 15.Gardner PS, McQuillin J. Applications of Immunofluorescence. 2. London: Butterworths; 1981. Rapid virus diagnosis; pp. 56–91. [Google Scholar]
- 16.Navikas V, Link J, Wahren B, Persson CH, Link H. Increased levels of interferon-gamma (IFN-γ), IL-4 and transforming growth factor-beta (TGF-β) mRNA expressing blood mononuclear cells in human HIV infection. Clin Exp Immunol. 1994;96:59–63. doi: 10.1111/j.1365-2249.1994.tb06230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agarwal R, Chaturvedi UC, Misra A, Kapoor S, Nagar R, Tandon R. CD4 positive T cells produce cytotoxic factor in cases of dengue haemorrhagic fever. Curr Sci. 1998;74:237–9. [Google Scholar]
- 18.Schauer U, Jung T, Krug N, Frew A. Measurement of intracellular cytokines. Immunol Today. 1996;17:305–6. doi: 10.1016/0167-5699(96)30020-0. [DOI] [PubMed] [Google Scholar]