

Abstract
We recently reported the positional cloning of a candidate gene for hereditary hemochromatosis called HFE. The gene product, a member of the major histocompatibility complex class I-like family, was found to have a mutation, Cys-282 → Tyr (C282Y), in 85% of patient chromosomes. This mutation eliminates the ability of HFE to associate with β2-microglobulin (β2m) and prevents cell-surface expression. A second mutation that has no effect on β2m association, H63D, was found in eight out of nine patients heterozygous for the C282Y mutant. In this report, we demonstrate in cultured 293 cells overexpressing wild-type or mutant HFE proteins that both the wild-type and H63D HFE proteins form stable complexes with the transferrin receptor (TfR). The C282Y mutation nearly completely prevents the association of the mutant HFE protein with the TfR. Studies on cell-associated transferrin at 37°C suggest that the overexpressed wild-type HFE protein decreases the affinity of the TfR for transferrin. The overexpressed H63D protein does not have this effect, providing the first direct evidence for a functional consequence of the H63D mutation. Addition of soluble wild-type HFE/β2m heterodimers to cultured cells also decreased the apparent affinity of the TfR for its ligand under steady-state conditions, both in 293 cells and in HeLa cells. Furthermore, at 4°C, the added soluble complex of HFE/β2m inhibited binding of transferrin to HeLa cell TfR in a concentration-dependent manner. Scatchard plots of these data indicate that the added heterodimer substantially reduced the affinity of TfR for transferrin. These results establish a molecular link between HFE and a key protein involved in iron transport, the TfR, and raise the possibility that alterations in this regulatory mechanism may play a role in the pathogenesis of hereditary hemochromatosis.
Hereditary hemochromatosis (HH) is an autosomal recessive disorder of iron metabolism and represents one of the most common inherited disorders in individuals of Northern European descent with an estimated carrier frequency between 1 in 8 and 1 in 10 (1–3). In patients with HH, excessive iron deposition in a variety of organs leads to multiorgan dysfunction (4, 5). Recently, we reported a mutation in a major histocompatibility complex (MHC) class I-like gene, initially called HLA-H (6) and now renamed HFE. Eighty-three percent of HH patient DNAs were found to be homozygous for this mutation, which consists of a single base transition of G to A and results in a change of Cys-282 → Tyr (C282Y). Subsequent reports have confirmed the high frequency of this founder mutation in other HH patients (7–9).
The HFE protein is similar to MHC class I family molecules. All four of the invariant cysteine residues that form disulfide bridges in the α2 and α3 domains of MHC class I protein are present in HFE. One of these conserved cysteine residues is altered by the C282Y mutation. Coimmunoprecipitation studies of cell lysates from human embryonic kidney cells transfected with wild-type or mutant HFE cDNA, demonstrate that wild-type HFE binds β2-microglobulin and that the C282Y mutation, completely abrogates this interaction (10, 11). Immunofluorescence and subcellular fractionations demonstrated that wild-type HFE is localized mainly on the cell surface whereas, the C282Y mutant protein is primarily intracellular (10, 11).
A second missense mutation, His-63 → Asp (H63D) has been reported to be enriched in hemochromatosis patients who are heterozygous for the C282Y mutation (6, 9). The functional consequence of this mutation has remained unclear. Recent studies demonstrated that the H63D mutation does not alter either the interaction with β2-microglobulin or the surface expression of the protein (10). By analogy to MHC class I structure, this mutation is located on the loop between the third and fourth β strands of the peptide-binding domain (12) and has been hypothesized to alter the manner in which HFE interacts with associating proteins or ligands (6).
The precise mechanism by which a MHC class I-like protein can influence iron homeostasis has remained obscure. To investigate this we utilized cell-surface labeling to detect potential HFE interacting proteins. We show that the HFE protein normally forms a stable complex with the transferrin receptor (TfR), the molecule responsible for receptor-mediated endocytosis of iron-bound transferrin (13–15). The consequence of this interaction is that the receptor exhibits a lower affinity for transferrin. The C282Y mutant protein forms only a trace amount of this complex, allowing high affinity transferrin binding to the uncomplexed TfR. By contrast, steady-state measurements suggested that the H63D mutant protein that does associate with the TfR has little influence on the apparent affinity of the TfR for transferrin. Taken together, these results provide a molecular link between HFE and iron metabolism and suggest how the major mutations may contribute to HH.
MATERIALS AND METHODS
Cell Surface Protein Biotinylations.
Construction of cell lines overexpressing wild-type and mutant forms of the FLAG epitope-tagged HFE protein have been previously described (10). Briefly, various HFE constructs cloned in pCDNA3.1 (Invitrogen) were introduced in 293 cells with LipofectAMINE (GIBCO) and clones isolated by selecting with 800 μg/ml of G418 (GIBCO). For surface biotinylations, cells (4 × 106) were seeded into 100-mm dishes and grown overnight to 80% confluency. The plates were moved to 4°C and gently washed four times with PBS. Sulfo-N-hydroxysuccinimide-LC biotin (Pierce) was added in PBS to a final concentration of 500 μg/ml and incubated on ice for 30 min. The biotin reagent was removed and the plates washed four times with PBS containing 50 mM glycine. Cells were lysed in 500 μl of 25 mM Tris⋅HCl, pH 7.5, 150 mM NaCl plus 0.5% Nonidet P-40. Protein concentrations were determined by BCA assay (Pierce) and 1 mg of protein was precleared with 25 μg of Protein-G-Sepharose (Pharmacia) and immunoprecipated with either 10 μg of anti-HFE rabbit polyclonal antibody (CT1) (10), 50 μg of FLAG (M2) mAb (Kodak), 5 μg of TfR monoclonal antibody (Caltag, South San Francisco, CA) or 10 μg of HLA-ABC antibody (Immunotech, Westbrook, ME). Precipitated proteins were separated on 4–20% Tris⋅glycine polyacrylamide gels (Novex, San Diego), electroblotted to polyvinylidene difluoride membranes (Novex) and biotinylated proteins were visualized with 2 μg/ml of streptavidin-horseradish peroxidase (Pierce) followed by enhanced chemiluminescence detection reagents (Amersham).
Immunoprecipitations and Western Blot Analysis.
Cells were lysed in the same buffer as above and precipitations carried out with the same antibodies and concentrations except that no preclearing step was carried out. Precipitated proteins were separated and electroblotted to polyvinylidene difluoride membranes as previously described (10). Gels were calibrated using prestained molecular weight standards (Novex).
Measurements of Cell-Associated Transferrin at Steady-State.
Measurements of cell-associated transferrin were carried out essentially as described (16) with the following modifications. Cells were seeded at a density of 6 × 105 per well in 6-well dishes coated with 0.01% fibronectin (Sigma) and grown overnight. Cells were washed once with 2 ml of DMEM-H21 (GIBCO) media supplemented with 1% fetal bovine serum and then incubated at either 37°C or on ice with varying concentrations of transferrin that include [125I]-diferric transferrin (1 μCi/μg; 1 Ci = 37 GBq) (NEN) as a tracer (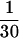 of the final concentration) in a final volume of 750 μl. To determine the amount of nonspecific transferrin binding, cells were simultaneously incubated under the same conditions but in the presence of 100× the molar concentration of cold holo-transferrin (Sigma). After 20 min (37°C), the media was removed and counted in a Beckman 9600 scintillation counter. The cells were placed on ice and washed two times with media containing 1% fetal bovine serum, and then lysed with 1% SDS and counted. Specific cell-associated transferrin was calculated by subtracting the nonspecific cell-associated transferrin from the total. A second method was also used that utilized a constant amount of labeled transferrin (5 nM) and increasing amount of unlabeled transferrin to increase the total transferrin concentration. Identical results to those produced by the first method were obtained. Transferrin binding in HeLa cells at 4°C was measured exactly as cell-associated transferrin except that the incubations were carried out at 4°C instead of 37°C and for 90 min instead of 20 min.
of the final concentration) in a final volume of 750 μl. To determine the amount of nonspecific transferrin binding, cells were simultaneously incubated under the same conditions but in the presence of 100× the molar concentration of cold holo-transferrin (Sigma). After 20 min (37°C), the media was removed and counted in a Beckman 9600 scintillation counter. The cells were placed on ice and washed two times with media containing 1% fetal bovine serum, and then lysed with 1% SDS and counted. Specific cell-associated transferrin was calculated by subtracting the nonspecific cell-associated transferrin from the total. A second method was also used that utilized a constant amount of labeled transferrin (5 nM) and increasing amount of unlabeled transferrin to increase the total transferrin concentration. Identical results to those produced by the first method were obtained. Transferrin binding in HeLa cells at 4°C was measured exactly as cell-associated transferrin except that the incubations were carried out at 4°C instead of 37°C and for 90 min instead of 20 min.
HeLa cells were grown to 80–90% confluence at 37°C at 5% CO2. The cells were washed twice with PBS and detached from the plates using 4 mM EDTA/PBS. The cells were counted and diluted to 5 × 106 cells per ml in DMEM 1% fetal bovine serum (Gibco) and 5 × 105 suspended cells were used for the TfR binding assay. Binding assays were all done in duplicate. Nonspecific binding was determined as for 293 cells. Soluble HFE was added to the cells for 10 min at room temperature prior to the addition of labeled transferrin where indicated. Labeled transferrin (NEN) was added to the cells at concentrations ranging from 0.5–15 nM. The reaction volume was 500 μL. Incubations were carried out for 20 min, for experiments performed at 37°C, or 90 min on ice for experiments performed at 4°C. At the end of the incubation the cells were pelleted at 3,000 rpm for 2 min. The supernatant was decanted and radioactivity counted. The cells were gently washed four times by resuspending the cells with 1 ml ice-cold DMEM 1% fetal bovine serum and repelleting at low centrifugal force. During the washing steps the cells were kept at 4°C. After the final wash, the cells were resuspended in PBS and bound radioactivity was counted. The amount of transferrin bound to the cells was calculated by dividing the cpm bound to the cells by the cpm counted in the supernatant, this ratio was converted to a molar value by multiplying by the amount of transferrin used in the experiment. The Kcell association and the apparent KD were calculated from the negative slope of the line generated by Scatchard analysis or by analyzing the binding curves utilizing nonlinear regression; both methods yielded essentially the same results.
Expression and Purification of Secreted HFE.
Secreted HFE was expressed in Chinese hamster ovary cells as follows: A 5′ XhoI site, a stop codon after the codon corresponding to amino acid 298 (6) (residue 276 of the mature protein) and a 3′ NotI site were inserted in the HFE gene by site-directed mutagenesis. After verifying the sequence, the modified HFE gene was subcloned into the expression vector PBJ5-GS that carries the glutamine synthetase gene (Lonza Biologics, Berkshire, U.K.) as a selectable marker and as a means of gene amplification in the presence of the drug methionine sulfoximine (17). The HFE expression plasmid was cotransfected with a human β2m expression vector (18) into Chinese hamster ovary K1 cells. Cell lines secreting HFE/β2m heterodimers were identified by immunoprecipitation of supernatants of [35S]-methionine metabolically labeled cells using an antibody against human β2m (BBM.1) (19). A protein of molecular mass of 43 kDa was coimmunoprecipitated with labeled β2m from the supernatants, and was verified to be the truncated HFE polypeptide chain by N-terminal sequencing of the purified protein (yielding the sequences RLLRSHSLHYLF and IQRTPKIQVYSR corresponding to the correctly processed mature forms of HFE and human β2m; data not shown). Soluble HFE/β2m heterodimers were purified on a BBM.1 immunoaffinity column, followed by separation of free β2m from the heterodimers on a Superdex 75 HR 10/30 fast protein liquid chromatography gel filtration column or by using an immunoaffinity column constructed with an HFE mAb raised against the purified heterodimer (J.A.L. and P.J.B., unpublished results).
RESULTS AND DISCUSSION
We initially utilized cell-surface labeling to detect potential HFE interacting proteins. Human embryonic kidney cells (293 cells), engineered to overexpress either wild-type or mutant forms of HFE, were treated with biotin-conjugated N-hydroxysuccinimide to label proteins expressed on the cell-surface. Subsequently, total cell lysates were immunoprecipitated with previously characterized antibodies directed toward the C-terminal peptide sequence of HFE or monoclonal antibodies against the FLAG epitope tag that had been engineered into the HFE protein (10). Biotinylated proteins were detected with streptavidin-conjugated horseradish peroxidase. Lysates from parental 293 cells displayed little surface-labeling in accordance with previous results demonstrating undetectable levels of HFE protein in these cells (Fig. 1 A and B) (10). In contrast, prominent bands at 49 and 100 kDa and the less intense bands at 12 and 200 kDa, were observed in immunoprecipitates from lysates of cells overexpressing the wild-type HFE; these bands were absent in immunoprecipitates from cells overexpressing the C282Y mutant form of HFE (Fig. 1 A and B). In previous studies it was demonstrated that the plasma membrane-bound form of HFE was 49 kDa in molecular mass and associated with β2m, a 12-kDa protein (10, 11). The presence of 49- and 12-kDa-labeled bands in HFE-specific immune-complexes from wild-type HFE expressing cells and their virtual absence in parental and C282Y mutant expressing cells is consistent with their identity as HFE and β2m. The fact that only trace amounts of the 100- and 200-kDa proteins coimmunoprecipitated from lysates of the C282Y mutant expressing cells suggests a specific interaction of these proteins with the cell-surface form of HFE.
Figure 1.

Cell-surface labeling of HFE and association with TfR. (A) HFE antibodies immunoprecipitate 12, 49, 100, and 200 kDa surface-labeled proteins from wild-type HFE expressing cells. The coimmunoprecipitating proteins are greatly reduced or absent in immune complexes from parental 293 or C282Y HFE mutant expressing cells. (B) FLAG epitope antibodies also immunoprecipitate 12, 49, 100, and 200 kDa surface-labeled proteins in wild-type HFE expressing cells. As in A, the coimmunoprecipitating proteins are greatly reduced or absent in immune complexes from parental 293 or C282Y HFE mutant expressing cells. (C) TfR antibodies immunoprecipitate 100- and 200-kDa surface-labeled proteins from parental 293, wild-type, and C282Y HFE expressing cells and in addition, immunoprecipitate 12- and 49-kDa proteins from wild-type HFE expressing cells. (D) HLA-ABC antibodies fail to immunoprecipitate 100- and 200-kDa proteins from parental 293 cells.
To further examine the specificity of the above protein interactions with HFE, we performed immunoprecipitations with antibodies that recognize the related HLA-A, B and C proteins. These antibodies detected proteins at ≈45 kDa and 12 kDa, the predicted molecular masses of the HLA heavy chain and β2m, but failed to coimmunoprecipitate the 100- and 200-kDa bands (Fig. 1D).
To identify the 100- and 200-kDa proteins that coimmunoprecipitated with HFE, we investigated proteins known to participate in iron homeostasis. Interestingly, the major carrier of transferrin-bound iron, the TfR is known to display a characteristic pattern of monomers and dimers migrating at ≈100 and 200 kDa in denaturing gel electrophoresis (13–15). To determine whether HFE could associate with the TfR, we utilized TfR antibodies to immunoprecipitate surface-labeled proteins from the three cell lines. A prominent protein of molecular mass corresponding to the monomeric TfR and a smaller amount of one corresponding to the dimeric form of the TfR were seen in the parental 293 as well as the wild-type HFE overexpressing cells. The C282Y mutant protein expressing cell line had less of both, (Fig. 1C). [However, studies of cell-associated transferrin suggest that these cells had equivalent numbers of receptors (see Fig. 3, compare A and B)]. Two proteins with masses corresponding to those of HFE and β2m (49 kDa and 12 kDa, respectively) were observed in immunoprecipitates from the cells that overexpress wild-type HFE protein. By contrast, little or no HFE protein was seen in immunoprecipitates from parental 293 or C282Y mutant protein expressing cells.
Figure 3.

Effect of HFE on cell-association of [125I]-transferrin. (A) Cell association of [125I]-transferrin in cells that overexpress the C282Y (intracellular) mutant form of HFE. Approximately 9 × 105 cells (clone 10, □; and clone 12, ▪) were incubated with various concentrations of transferrin at 37°C for 20 min (Inset). The data represent the mean of duplicate determinations corrected for nonspecific binding. Scatchard analysis revealed an Kcell association of ≈12 and 14 nM, respectively. (B) Cell-association of [125I]-transferrin in cells overexpressing the wild-type (surface) form of HFE (clone 7, ○; clone 3, •). Scatchard analysis revealed that the Kcell association for transferrin was 180 and 40 nM, respectively. (C) Cell-association of [125I]-transferrin in cells overexpressing the H63D mutant form of HFE (clone 3, ▵; clone 8, ▴). Scatchard analysis revealed a Kcell association for transferrin to be 12 and 20 nM, respectively. (D) Inhibition of cell-associated [125I]-transferrin to the TfR on parental 293 cells by addition of soluble HFE/β2m heterodimers. Parental 293 cells were preincubated with various amounts of soluble HFE/β2m heterodimers followed by addition of 10 nM [125I]-transferrin. The amount of specific cell-associated transferrin was determined and plotted as a percent of that observed in cells with no added HFE. The apparent Ki is 87 nM.
The HFE/TfR association results were corroborated by performing coimmunoprecipitation experiments on unlabeled total cell lysates. Immunoprecipitation with HFE antibodies followed by blotting and probing with antibodies to TfR demonstrated that the TfR was complexed with the wild-type form of HFE. In contrast, very little TfR was observed in the immunoprecipitates from the C282Y mutant cells (Fig. 2A). Reprobing blots with the FLAG epitope antibodies to detect HFE demonstrated that equivalent amounts of HFE were being expressed and immunoprecipitated from each of the cell lines except the parental 293 cells, as expected (Fig. 2B). Performing the inverse experiment, wherein cell lysates were first immunoprecipitated with TfR antibodies followed by blotting with HFE antibodies, revealed that HFE coimmunoprecipitated with the TfR from wild-type HFE expressing cells; only a trace band was observed with C282Y and none with the parental 293 cell line (Fig. 2C). The absence of HFE in immunoprecipitates with TfR in parental 293 and near absence in the C282Y mutant cell lines was not due to failure to precipitate TfR; reprobing the blot with TfR antibodies demonstrated that TfR protein was precipitated from each of the cell lines, including C282Y (Fig. 2D). To further control for the specificity of the HFE antibodies, we first immunoprecipitated cell lysates with FLAG epitope antibodies to specifically precipitate the HFE/FLAG fusion proteins followed by blotting with TfR antibodies. As was observed in Fig. 2A, the TfR was coimmunoprecipitated from cells expressing the FLAG-tagged wild-type HFE protein, but not from cells expressing the FLAG-tagged C282Y mutant HFE protein (Fig. 2E). Experiments performed on an independent series of cell lines engineered to express wild-type and mutant HFE that lacked the FLAG epitope tag yielded identical results to those shown in Fig. 2 when immunoprecipitations were carried out with HFE and TfR antibodies (data not shown).
Figure 2.

Direct association of TfR with HFE. (A) HFE antibodies coimmunoprecipitate TfR from wild-type and H63D HFE expressing cells but very little from parental 293 or C282Y HFE mutant expressing cells. (B) HFE antibodies immunoprecipitate substantial amounts of HFE protein from wild-type, C282Y and H63D HFE expressing cells. (C) TfR antibodies coimmunoprecipitate HFE from wild-type and H63D HFE expressor cells but not parental 293 and only a trace from C282Y mutant expressing cells. (D) TfR antibodies immunoprecipitate TfR protein from parental 293, and wild-type, C282Y and H63D HFE expressing cells. (E) FLAG epitope (M2) antibodies coimmunoprecipitate TfR from wild-type and H63D HFE expressing cells but not parental 293 or C282Y HFE mutant expressing cells.
In immunoprecipitation experiments on unlabeled cell lysates we included as a further control another mutant of HFE wherein His-63 was replaced by aspartate (H63D). As with wild-type HFE, the H63D protein is also expressed on the cell surface and associates with β2m (10, 11). The association of HFE with the TfR as assessed by coimmunoprecipitation appeared unaffected by the H63D mutation (Fig. 2 A, C, and E).
To assess the biological effect of the HFE/TfR interaction, we characterized the transferrin-binding properties of the TfR in the presence or absence of HFE. For these studies we examined the amount of [125I]-transferrin (diferric form) associated with intact 293 cells engineered to overexpress both β2m and either wild-type HFE or one of two HFE mutant forms, C282Y or H63D. The initial binding experiments were performed at 37°C under which conditions the total cell-associated [125I]-transferrin represents both surface-bound and internalized ligand at steady-state (20, 21). The cell-association of [125I]-transferrin saturated at 150–250 nM on both C282Y mutant and wild-type HFE expressing cells (Fig. 3 A and B insets, which present data from two separate cell clones for each the wild-type and mutant HFE). When subjected to Scatchard analysis, the C282Y HFE mutant expressing clones showed a Kcell association for transferrin of ≈12 and 14 nM (Fig. 3A). A similar Kcell association for transferrin was found in parental 293 cells (19 nM) (data not shown). By contrast, the Kcell association for transferrin in the wild-type HFE expressing clones, appeared to be increased 3- and 15-fold to Kcell association values of 40 and 180 nM, respectively.
In contrast to the effect of wild-type HFE on Kcell association for transferrin, the H63D mutation, while retaining the ability to interact with the TfR (Figs. 1 and 2), did not appear to increase the Kcell association for transferrin. Two clonal lines expressing the H63D mutant protein had apparent Kcell association for transferrin of ≈12 and 20 nM (Fig. 3C). These values are nearly identical to those obtained with the cell lines expressing the C282Y mutant in which the HFE protein was absent from the cell surface (Kcell association = 12 and 14 nM). The total amount of cell-associated transferrin in cell lines expressing the H63D mutant (1. 8 × 10−10 M) was 2 to 3-fold lower when compared with the C282Y expressing cells (4–6 × 10−10 M) (Fig. 3 A and C). This may reflect clonal variation or may reflect an altered TfR trafficking. Regardless, these data suggest that one functional consequence of the H63D mutation appears to be the loss of the ability of the HFE protein to increase the Kcell association for transferrin. In lacking this property of the wild-type HFE protein, HFE with the H63D mutation functionally resembles the C282Y mutation.
As an alternative method to assess the effect of HFE on the Kcell association for transferrin, we added a soluble form of HFE/β2m heterodimers to the culture medium of parental 293 cells. The addition of the HFE/β2m heterodimers at 37°C reduced cell-associated transferrin in a concentration-dependent manner with maximal inhibition at 2 μM heterodimer (Fig. 3D). The apparent Ki for inhibition was 87 nM. Similarly, in the presence of 2 μM soluble HFE/β2m heterodimers the Kcell association for transferrin was increased 5-fold to 100 nM (data not shown). Control experiments using an identical amount of an MHC class I, H-2Kd (22) protein complexed with human β2m failed to have any effect on the cell-associated transferrin to 293 cells (data not shown).
To corroborate the inhibitory effects observed with soluble HFE/β2m heterodimers on 293 cells we performed similar studies on HeLa cells. Scatchard analysis of the cell-associated transferrin data obtained for HeLa cells at 37°C revealed an apparent Kcell association of 4.7 nM (Fig. 4A). In the presence of either 150 nM or 1 μM soluble HFE/β2m heterodimers, the apparent Kcell association increased to 11 nM or 50 nM, respectively (Fig. 4A). An apparent Ki for inhibition of cell-associated transferrin for the soluble HFE/β2m heterodimers was calculated to be 39 nM (Fig. 4B). To determine whether the increase in the Kcell association for transferrin was specific for the HFE protein, we added to the assay a classical MHC class I protein, a purified soluble form of HLA-B27, complexed with β2m (23). Addition of 100 nM HLA-B27 had no effect on the Kcell association of transferrin binding to the TfR (Fig. 4A), demonstrating that the inhibitory effect of the HFE/β2m heterodimers was specific to HFE, and was not attributable solely to the presence of excess amounts of MHC class I molecules in the assay.
Figure 4.

Soluble HFE/β2m heterodimers reduce the affinity of the TfR for transferrin. (A) Effect of HFE/β2m heterodimers on the cell-association of [125I]-transferrin to HeLa cells at 37°C. Cell-associated [125I]-transferrin in HeLa cells in the presence of 0, 150, or 1,000 nM HFE or 100 nM soluble HLA-B27/β2m heterodimers (Inset). Scatchard analysis of cell associated [125I]-transferrin in the presence of 0 nM (□), 150 nM (▴), or 1,000 nM HFE (▵) revealed Kcell association values of 4.7 nM, 11 nM, and 50 nM, respectively and ≈1.0 × 106 transferrin binding sites per cell. (Note: the addition of 1,000 nM soluble HFE/β2m heterodimers dramatically reduces cell-associated transferrin such that an accurate x-intercept is difficult to obtain; the Kcell association 50 nM is estimated utilizing 1.0 × 106 binding sites per cell.) Addition of 100 nM soluble HLA-B27/β2m heterodimers (▪) to the assay had no effect. (B) Determination of the apparent Ki of soluble HFE/β2m heterodimers for cell-associated transferrin inhibition in HeLa cells. Cells were preincubated at 37°C in various amounts of soluble HFE/β2m heterodimers and then [125I]-transferrin was added to 5 nM. The amount of specific cell-associated transferrin was determined and plotted as a percent of that observed in cells with no added HFE. The apparent Ki for HeLa is 39 nM. (C) Effect of HFE/β2m heterodimers on the binding of [125I]-transferrin to HeLa cells at 4°C. Binding of [125I]-transferrin to HeLa cells in the absence or presence of 0, 35, or 1,000 nM HFE (inset). Scatchard analysis of [125I]-transferrin binding in the presence of 0 (□), 35 (▴), or 1,000 nM (▵) HFE revealed apparent KD values of 5 nM, 12 nM, and 75 nM, respectively and ≈1.0 × 106 transferrin binding sites per cell. (Note: the addition of 1,000 nM soluble HFE/β2m heterodimers dramatically reduces transferrin binding such that an accurate x intercept is difficult to obtain; the apparent KD of 75 nM is estimated utilizing 1.0 × 106 binding sites per cell.)
The transferrin binding studies were repeated at 4°C where internalization of ligand is negligible, to determine whether the observed effects of the soluble HFE/β2m heterodimers were due to changes in the KD of the TfR for transferrin (24–26). We utilized HeLa cells for these studies because of the high nonspecific transferrin binding in 293 cells observed at 4°C. Scatchard analysis of the binding data revealed that in the absence of HFE, the TfR bound transferrin with an apparent KD of 5 nM, which was similar to the Kcell association of 4.2 nM observed at 37°C (Fig. 4C). In the presence of either 35nM or 1 μM soluble HFE/β2m heterodimers the apparent KD increased to 12 and 75 nM, respectively, indicating that the principal effect of added HFE/β2m heterodimers is to reduce the affinity of the TfR for transferrin. Further, as was the case for Kcell association at 37°C, addition of 100 nM soluble HLA-B27/β2m heterodimers to the assay had no effect on transferrin binding at 4°C, supporting the specificity of the inhibition by HFE (data not shown). These experiments demonstrate that wild-type HFE can lower the affinity of TfR for transferrin.
The primary defects in hereditary hemochromatosis appear to be both increased iron absorption in the small intestine and increased iron deposition in major organs (4, 5). We have demonstrated here that HFE forms a stable complex with the TfR and decreases the affinity of transferrin binding. The C282Y mutation largely eliminates this interaction. We have also shown that the second missense mutation associated with hemochromatosis, H63D, appears to have a similar functional consequence in that it alters the ability of HFE to increase the Kcell association for transferrin. The loss of HFE-repressor function for transferrin uptake could result in increased cellular uptake of iron by some tissues, and contribute to iron deposition in HH. How these mutations lead to increased iron absorption in the intestine is unclear. Recent immunohistochemical studies have localized HFE to the intracellular portion of the cells in the deep crypts of the duodenum (27), the same region where previous studies have localized the TfR (28, 29). The role of the TfR in the cells of the deep crypts has long been thought to be limited to servicing the proliferative needs of these cells. In light of the association of HFE and the TfR, one must now reconsider the role of transferrin and its receptor in regulating intestinal iron absorption. Regardless of the actual mechanism, the observations described here provide a molecular link between HFE and iron metabolism and raise the possibility that perturbation of the transferrin/TfR system of iron transport is a previously unrecognized component of HH disease. Furthermore, these results demonstrated that by administering HFE protein in situ, the amount of transferrin taken up by cells can be attenuated. Possibly the HFE/TfR interaction provides a new target for therapeutic agents aimed at influencing iron absorption and/or tissue distribution in iron overload disorders.
Acknowledgments
We thank C. Enns, A. Grinke, and W. Thomas for scientific discussion and critical reading of the manuscript, and N. Moeller for her assistance in tissue culture. J.A.L. was supported by a FORD fellowship.
ABBREVIATIONS
- HH
hereditary hemochromatosis
- β2m
beta-2 microglobulin
- TfR
transferrin receptor
References
- 1.Dadone M M, Kushner J P, Edwards C Q, Bishop D T, Skolnick M H. Am J Clin Pathol. 1982;78:196–207. doi: 10.1093/ajcp/78.2.196. [DOI] [PubMed] [Google Scholar]
- 2.Edwards C Q, Griffin L M, Goldgar D, Drummond C, Skolnicx M H, Kushner J P. N Engl J Med. 1988;318:1355–1362. doi: 10.1056/NEJM198805263182103. [DOI] [PubMed] [Google Scholar]
- 3.McLaren C E, Gordeuk V R, Looker A C, Hasselblad V, Edward C Q, Griffen L M, Kushner J P, Brittenham G M. Blood. 1995;86:2021–2027. [PubMed] [Google Scholar]
- 4.Bothwell T H, Charlton R W, Motulsky A G. In: The Metabolic and Molecular Basis of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw-Hill; 1995. pp. 2237–2269. [Google Scholar]
- 5.Bacon B R, Tavill A S. In: Hepatology. A Textbook of Liver Disease. Zakim D, Boyer T D, editors. Philadelphia: Saunders; 1996. pp. 1439–1472. [Google Scholar]
- 6.Feder J N, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy D A, Basava A, Dormishian F, Domingo R J, Ellis M C, Fullan A, et al. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 7.Jazwinska E C, Cullen L M, Busfield F, Pyper W R, Webb S I, Powell L W, Morris C P, Walsh T P. Nat Genet. 1996;14:250–251. doi: 10.1038/ng1196-249. [DOI] [PubMed] [Google Scholar]
- 8.Jouanolle A M, Gandon G, Jezequel P, M, B, Campion M L, Yaouanq J, Mosser J, Fergelot P, Chauvel B, Bouric P, Carn G, et al. Nat Genet. 1996;14:251–252. doi: 10.1038/ng1196-251. [DOI] [PubMed] [Google Scholar]
- 9.Beutler E, Gelbart T, West C, Lee P, Adams M, Blackstone R, Pockros P, Kosty M, Venditti C P, Phatak P D, et al. Blood Cells Mol Dis. 1996;31:187–194. doi: 10.1006/bcmd.1996.0027. [DOI] [PubMed] [Google Scholar]
- 10.Feder J N, Tsuchihashi Z, Irrinki A, Lee V K, Mapa F A, Morikang E, Prass C E, Starnes S M, Wolff R K, Parkkila S, et al. J Biol Chem. 1997;272:14025–14028. doi: 10.1074/jbc.272.22.14025. [DOI] [PubMed] [Google Scholar]
- 11.Waheed A, Parkkila S, Zhou X Y, Tomatsu S, Tsuchihashi Z, Feder J N, Schatzman R C, Britton R S, Bacon B R, Sly W S. Proc Natl Acad Sci USA. 1997;94:12384–12389. doi: 10.1073/pnas.94.23.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjorkman P J, Saper M A, Samraoui B, Bennett W S, Strominger J L, Wiley D C. Nature (London) 1987;329:506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- 13.Seligman P A, Schleicher R B, Allen R H. J Biol Chem. 1979;254:9943–9946. [PubMed] [Google Scholar]
- 14.Wada H G, Hass P E, Sussman H H. J Biol Chem. 1979;254:12629–12635. [PubMed] [Google Scholar]
- 15.Omary M B, Trowbridge I S. J Biol Chem. 1981;256:12888–12892. [PubMed] [Google Scholar]
- 16.Ward J H, Kushner J P, Kaplan J. J Biol Chem. 1982;257:10317–10323. [PubMed] [Google Scholar]
- 17.Bebbingtion C R, Hentschel C G G. In: DNA Cloning: A Practical Approach. Glover D M, editor. Oxford: IRL; 1987. pp. 163–188. [Google Scholar]
- 18.Fahnestock M L, Johnson J L, Feldman R M R, Neveu J M, Lane W S, Bjorkman P J. Immunity. 1995;3:583–590. doi: 10.1016/1074-7613(95)90129-9. [DOI] [PubMed] [Google Scholar]
- 19.Parham P, Androlewicz M J, Holmes N J, Rothenberg B E. J Biol Chem. 1983;258:6179–6186. [PubMed] [Google Scholar]
- 20.Karin M, Mintz B. J Biol Chem. 1981;256:3245–3252. [PubMed] [Google Scholar]
- 21.Octave J N, Schneider Y J, Hoffmann P, Trouet A, Crichton R R. Eur J Biochem. 1982;123:235–240. doi: 10.1111/j.1432-1033.1982.tb19758.x. [DOI] [PubMed] [Google Scholar]
- 22.Fahnestock M L, Tamir I, Narhi L, Bjorkman P J. Science. 1992;258:1658–1662. doi: 10.1126/science.1360705. [DOI] [PubMed] [Google Scholar]
- 23.Raghaven M, Lebron J A, Johnson J L, Bjorkman P J. Protein Sci. 1996;5:2080–2088. doi: 10.1002/pro.5560051014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weigel P H, Oka J A. J Biol Chem. 1982;257:1201–1207. [PubMed] [Google Scholar]
- 25.Klausner R D, Renswoude J V, Ashwell G, Kempf C, Schechter A N, Dean A, Bridges K R. J Biol Chem. 1983;258:4715–4724. [PubMed] [Google Scholar]
- 26.Mulford C A, Lodish H F. J Biol Chem. 1988;263:5455–5461. [PubMed] [Google Scholar]
- 27.Parkkila S, Waheed A, Britton R S, Feder J N, Tsuchihashi Z, Schatzman R C, Bacon B R, Sly W S. Proc Natl Acad Sci USA. 1997;94:2534–2539. doi: 10.1073/pnas.94.6.2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banerjee D B, Flanagan P R, Cluett J, Valberg L S. Gastroenterology. 1986;91:861–869. doi: 10.1016/0016-5085(86)90687-6. [DOI] [PubMed] [Google Scholar]
- 29.Anderson G J, Powell L W, Halliday J W. Gastroenterology. 1990;98:576–585. doi: 10.1016/0016-5085(90)90276-7. [DOI] [PubMed] [Google Scholar]