

Abstract
Pharmacologic glucocorticoids are powerful inhibitors of the inflammatory response at many levels, including leucocyte trafficking and function. The adhesion molecule P-selectin is a key participant in polymorphonuclear neutrophil (PMN) migration to sites of inflammation. The extent to which endogenous glucocorticoids influence PMN migration and activation is not clear. We used the glucocorticoid receptor antagonist RU486 to examine the effect of endogenous glucocorticoid blockade on PMN migration and function in carrageenan monoarthritis in the rat. Arthritis was induced by intra-articular injection of carrageenan and disease severity measured by PMN count in synovial lavage fluid. Decalcified frozen sections of injected joints were analysed for expression of P-selectin by immunohistochemistry. Adrenal glucocorticoid action was blocked in vivo with RU486 20 mg/kg. PMN phagocytosis and reactive oxygen species synthesis were measured by flow cytometry. Carrageenan injection was associated with severe arthritis (synovial lavage PMN 5.9 ± 0.7 × 106, P < 0.01 versus control) which was dose-dependent. P-selectin was not detected in normal joints but was abundant in joints injected with 500 μg carrageenan. RU486 resulted in exacerbation of carrageenan arthritis (9.7 ± 0.8 × 106, P < 0.05). RU486 also altered the threshold for disease induction, in that most RU486-treated animals were susceptible to arthritis at a dose of carrageenan (2.5 μg) which did not induce arthritis in most control-treated animals (P < 0.05), denoting an altered threshold for arthritis induction. RU486 treatment was associated with increased synovial P-selectin expression. Activation status as measured by PMN phagocytic and oxidative function were not influenced by endogenous glucocorticoid blockade. These findings suggest that endogenous glucocorticoids selectively influence PMN migration to inflamed joints via P-selectin expression, but have no effect on PMN activation status.
Keywords: neutrophil, P-selectin, synovium, glucocorticoids, RU486
INTRODUCTION
The anti-inflammatory effects of glucocorticoids have been extensively described in experimental models of inflammatory disease, and their efficacy has been borne out in therapeutic situations in human disease. The concept that glucocorticoids released under physiological conditions contribute to an endogenous inflammatory control system is now well accepted. For example, the limitation of severity and duration of inflammatory disease models including adjuvant arthritis and experimental allergic encephalomyelitis (EAE) has been demonstrated to be dependent on adequate physiologic glucocorticoid responses [1,2]. Furthermore, the studies of Sternberg et al. [3] demonstrate that replacement of glucocorticoids in the physiologic range ameliorates streptococcal cell wall-induced arthritis in Lewis rats, whose susceptibility to disease is in turn dependent upon impaired glucocorticoid production. In human rheumatoid arthritis (RA), disease activity has been correlated inversely with adrenal cortisol secretion [4], and blockade of cortisol production leads to exacerbation of clinical symptoms [5]. At the cellular level, there is evidence for regulation of inflammatory events including natural killer (NK) cytotoxicity, nitric oxide production, T cell activation, expansion and programmed cell death in vivo by endogenous glucocorticoids [6–9].
In RA, large numbers of polymorphonuclear neutrophils (PMN) are found in the synovial fluid of inflamed joints. We have recently reported the role of PMN in the evolution of rat adjuvant arthritis model of human RA [10], and the importance of these cells to inflammatory joint disease is increasingly recognized [11]. PMN recruitment to the joint is known to be dependent upon expression of PMN adhesion molecules including P- and E-selectin, and the production of chemotactic factors such as IL-8 [12–16]. Reactive oxygen species (ROS) and proteolytic enzymes derived from activated PMN may be involved in initiation or perpetuation of joint damage, as well as recruitment of other inflammatory cells. It has been recently shown that administration of pulse methylprednisolone resulted in a significant decrease in synovial fluid PMN in RA [17,18], and that glucocorticoids are associated with reductions in IL-8 production [19]. In contrast, the effects of endogenous glucocorticoids on synovial PMN recruitment have not been studied. Furthermore, although exogenous glucocorticoids are known to influence the expression of key adhesion molecules involved in PMN recruitment [20], the impact of endogenous glucocorticoids on P-selectin expression has not been studied. The potential for the immunomodulatory effects of physiologic glucocorticoids to differ markedly from those of pharmacologic doses has recently been highlighted [21]. We therefore used a model of PMN-dependent arthritis to investigate endogenous glucocorticoid effects on acute PMN-mediated synovitis.
MATERIALS AND METHODS
Animals and interventions
Adult male inbred Sprague-Dawley rats weighing 300–350 g were housed six to a cage and maintained on a 12:12 h light:dark cycle with standard rat chow and water ad libitum. RU486 (a gift from Roussel-UCLAF, Roumainville, France) was prepared as a suspension in saline containing 0.5% carboxymethyl cellulose (Sigma, St Louis, MO). Carrageenan was prepared as a 1% stock solution in 0.9% NaCl. When lower concentrations were required for threshold experiments, carrageenan was dissolved in sterile 0.5% carboxymethyl cellulose in saline to maintain viscosity. RU486 (10 mg) was administered to rats 18 h and 1 h prior to arthritis induction, via i.p. injection. Control rats received i.p. injections of vehicle. Arthritis was induced by intra-articular (i.a.) injection of carrageenan (0.5–500 μg in 50 μl) into knee joints under ether anaesthesia. Inflammatory arthritis (defined by a synovial lavage PMN count > 0.5 × 106) developed within 4 h. Animals were killed at this time and joints lavaged with 5 ml of 0.9% NaCl. Experiments were approved by the Research Ethics Committee of Monash University. Except where otherwise stated, six animals were examined for each data point.
Determination of anti-glucocorticoid effects
PMN number
Joint lavage samples were washed in PBS and cells were counted after erythrocyte lysis. Leucocytes which were identified as 100% PMN by light microscopic determination of characteristic nuclear morphology were counted in a Nebauer haemocytometer.
PMN function
Synovial lavage PMN activation was assessed in terms of phagocytic and oxidative function [22,23]. Phagocytosis and subsequent ROS generation were stimulated by opsonized propidium iodide (PI)-labelled Staphylococcus aureus Cowan (Pansorbin) (Calbiochem, San Diego, CA). PI (100 μg/ml) was incubated with an equal volume of Pansorbin for 30 min at room temperature. After washing in Hanks' balanced salt solution (HBSS), the bacteria were opsonized by incubation with 10% normal rat serum in HBSS at 37°C for 30 min, counted, and resuspended in HBSS at 0.5 × 106 cells/ml. Pansorbin-PI (50 μl) was added to 500 μl of the cell suspension and incubated at 37°C for 50 min. Dihydrorhodamine (DHR)-123 (10 μl; 20 mg/ml in PBS) was added and incubated for a further 10 min at 37°C. Samples were then placed on ice and flow cytometric analysis performed within 10 min. Phagocytosis was detected by flow cytometric measurement of PI-fluorescent cells. ROS production was measured by flow cytometric analysis of DHR-123 fluorescent cells (Coulter, Hialeah FL). Oxidation of the non-fluorescent DHR to green fluorescent rhodamine is initiated preferentially by H2O2 generation which occurs distal to NADPH-oxidase catalysed superoxide production.
An alternative method of inducing ROS production was employed using the calcium ionophore A23187. A23187 (Sigma) dissolved in dimethylsulphoxide was added at a final concentration of 1 mm to joint lavage specimens washed as before and resuspended in calcium-enriched PBS (Dulbecco B) containing 10 μm sodium azide. ROS production was measured by mean fluorescence intensity (MFI) of DHR-123-fluorescent cells.
Immunohistochemistry
Specimens of whole rat knee joint were fixed in PLP and decalcified in a solution of 7.5% polyvinylpyrrolidone (Sigma, Australia) and 10% ethylenediamine tetra acetic acid (EDTA; BDH Chemicals, Australia). Decalcified joints were embedded in OCT (Tissue Tek, Westhaven, CT) and frozen. Frozen tissue was cut into 7-μm sections using a cryostat (Reichardt-Jung Cryocut 1800; Germany). Immunohistochemistry was performed using a three-layer immunoperoxidase technique on sections from whole decalcified rat joints. P-selectin was detected using a rabbit anti-rat P-selectin polyclonal antibody as published [24], followed by peroxidase-conjugated goat anti-mouse antibody (Dako Corp., Carpinteria, CA) and mouse peroxidase anti-peroxidase 1:100 (Dako). Immunostaining was achieved by the addition of diaminobenzidine (Dako; 100 μg/ml). Sections were counterstained with Harris haematoxylin (Gurr BDH Chemicals, UK) prior to examination.
Statistical analysis
Statistical comparisons between groups were made using Student's t-test. Disease threshold data were analysed using χ2 analysis with continuity correction. All results are presented as mean ± s.e.m.
RESULTS
Induction of monoarthritis and P-selectin expression
Intra-articular administration of 0.5% carboxymethyl cellulose did not produce arthritis (synovial PMN = 0, n = 4). Intra-articular administration of carrageenan (500 μg) induced arthritis in 98% of animals (synovial lavage PMN 5.9 ± 0.7 × 106, P = 0.001 compared with control, n = 20). Lower doses induced PMN infiltration in a dose-dependent manner (Fig. 1). Compared with control-injected joints, histological examination of carrageenan-injected joints revealed intense infiltration with PMN in the synovial membrane (Fig. 2). Using immunohistochemical labelling, P-selectin was not detected in control-injected joints, but intense staining was detected in synovial blood vessels in carrageenan-injected joints (Fig. 2).
Fig. 1.
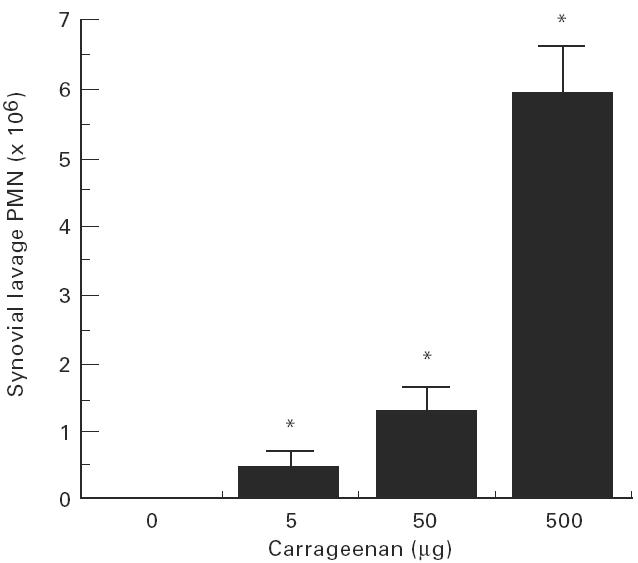
Rat knee joints were injected with vehicle (0.5% carboxymethyl cellulose in saline) or carrageenan (5–500 μg) and synovial lavage leucocytes counted after 4 h. Carrageenan induced a significant (*P < 0.01) and dose-dependent influx of polymorphonuclear neutrophils (PMN) to the synovial space.
Fig. 2.
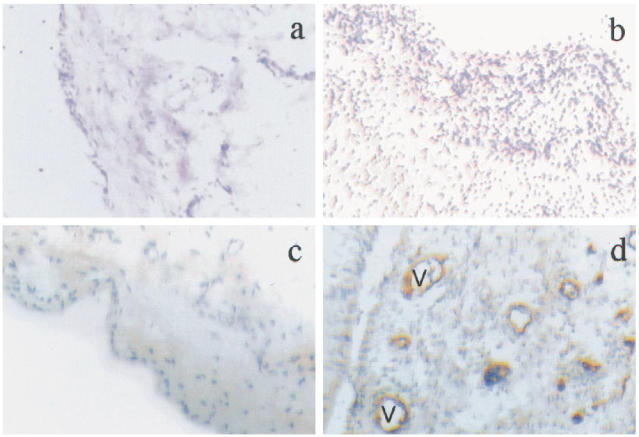
Rat knee joints were injected with vehicle (0.5% carboxymethyl cellulose in saline) or carrageenan (500 μg) and stained with haematoxylin and eosin (H–E) for histological assessment, or with anti-P-selectin antibody (brown) and haematoxylin. (Mag. × 100.) (a) Vehicle-injected (H–E). (b) Carrageenan-injected rat knee joint (H–E), showing marked increase in synovial leucocyte infiltration. (c) Vehicle-injected rat knee joint stained for P-selectin, showing negative staining. (d) Carrageenan-injected rat knee joint stained for P-selectin, showing positive brown staining in synovial blood vessels (V).
Endogenous glucocorticoid modulation of arthritis severity and threshold
Pretreatment with RU486 resulted in exacerbation of arthritis induced by 500 μg carrageenan (synovial lavage PMN 9.7 ± 0.8 × 106, P = 0.001, n = 20). At all doses of carrageenan able to induce arthritis in control animals, treatment with RU486 was associated with increased severity of arthritis (Table 1). An arthritogenic threshold dose of carrageenan, defined as a dose inducing arthritis in < 20% of animals, was observed at 2.5 μg (Table 2). In contrast to control-treated animals, > 50% of RU486-treated animals developed arthritis at this dose (Table 2). P-selectin was not detected in joints injected with 2.5 μg carrageenan in control-treated animals (Fig. 3). In contrast, RU486 treatment was associated with the intense expression of synovial P-selectin at this dose of carrageenan (Fig. 3).
Table 1.
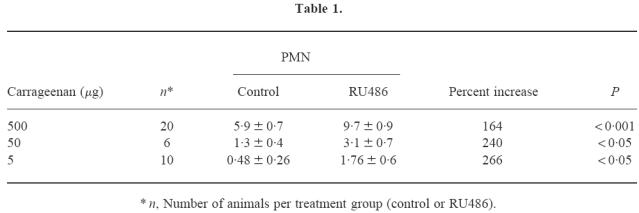
Table 2.
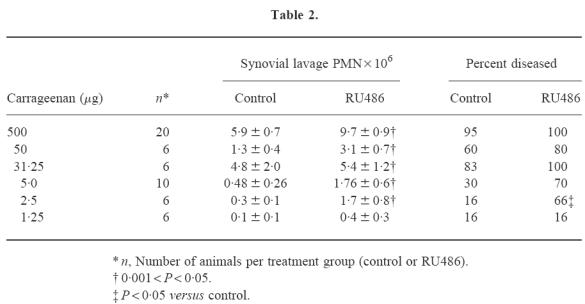
Fig. 3.
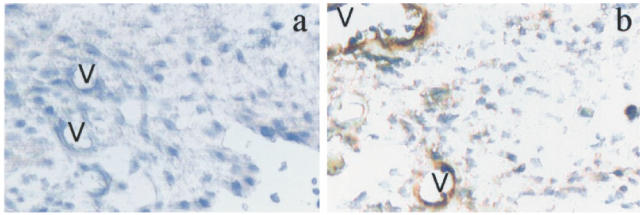
Rat knee joints were injected with carrageenan (2.5 μg) and stained with anti-P-selectin antibody (mag. × 200). (a) Control-treated rats exhibited no P-selectin in synovial blood vessels (V). (b) RU486-treated rats exhibited prominent P-selectin in synovial blood vessels (V).
Endogenous glucocorticoid modulation of PMN activation
Phagocytic capacity and ROS generation were detected in synovial lavage PMN of 100% of arthritic animals. Synovial lavage PMN phagocytic function as measured by flow cytometry (MFI 66.1 ± 8.2) was not influenced by blockade of endogenous glucocorticoids (65.4 ± 10.1, NS). ROS generation induced by bacterial phagocytosis (MFI of DHR 46.1 ± 9.7) was also not affected by RU486 treatment in vivo (43.6 ± 7.6, NS). Similarly, ROS generation induced by A23187 stimulation (102.2 ± 19.3) was not influenced by RU486 (100.5 ± 19.8, NS).
DISCUSSION
Several lines of evidence implicate PMN in inflammatory joint disease. For example, MoAb-mediated depletion of PMN is associated with significant inhibition of adjuvant arthritis in rats [10], human RA synovial fibroblast adhesion to human cartilage is enhanced by pretreatment with PMN proteases [25], and a correlation has been reported between PMN activation and the development of erosions in early RA [26]. Moreover, extensive production of cytokines by human PMN in vitro and in vivo suggests that PMN may regulate cytokine production by other inflammatory cells [27,28]. Although the concept that endogenous glucocorticoids contribute to regulation of the inflammatory response is widely accepted, PMN-mediated inflammatory models and PMN functions in vivo are possibly the least well understood aspect of the immunomodulatory role of physiologic glucocorticoids. The current study addresses the role of endogenous glucocorticoids in the early phase of carrageenan arthritis, which involves infiltration of PMN into the synovial fluid and membrane. We report that synovial PMN infiltration is significantly exacerbated by RU486 over a range of doses. Although the mechanisms of endogenous glucocorticoid modulation of human RA [5] is not understood, exogenous glucocorticoids have been reported to influence synovial PMN migration in human RA [17,18]. Endogenous glucocorticoids influence adjuvant arthritis [1], a model of RA known to be dependent on the role of PMN [10] and on P-selectin [29]. Taken together, this information strongly suggests that endogenous glucocorticoids may modulate human RA by mechanisms including P-selectin-mediated PMN migration.
The current study also demonstrates that endogenous glucocorticoids also modulate the threshold for disease development. Few studies have addressed the role of endogenous glucocorticoids in determining the threshold for disease development or susceptibility to inflammatory disease. Susceptibility to streptococcal cell wall (SCW)-induced arthritis in HPA-deficient Lewis rats is reversed by physiologic replacement of glucocorticoids [3], and susceptibility to EAE in the normally resistant PVG rat is induced by adrenalectomy [2]. In these two studies, however, the susceptibility of different rat strains, rather than a dose threshold for disease development, was examined. There has been no previous investigation of the role of physiologic glucocorticoids in determining the threshold for development of joint inflammation in a dose-dependent disease model. This finding is of potential importance, given the evidence for impaired HPA axis function in humans with RA [30,31] and the postulated role of antigen-driven immune responses in this disease.
The mechanism of increased PMN numbers in inflamed joints of RU486-treated animals necessarily involves increased transmigration across vascular endothelium by PMN. From studies of P-selectin gene-knockout mice, it is clear that this molecule has a crucial role in PMN rolling and extravasation [32]. Moreover, it has recently become clear that P-selectin is an important contributor to leucocyte migration in human RA and animal models [14,15]. Our finding that synovial P-selectin is negative in control joints and is detected in arthritic joints strongly suggests that P-selectin is an important contributor to the evolution of this model. Furthermore, the observation that P-selectin is influenced by endogenous glucocorticoids suggests that alterations in P-selectin contribute to increases in synovial PMN observed with glucocorticoid blockade. The current study is the first in an inflammatory setting, although Suzuki et al. reported increased leucocyte adherence and P-selectin-mediated leucocyte rolling response in RU486-treated hypertensive rats [33]. Additional mechanisms may influence PMN migration to inflammatory sites in the context of glucocorticoid blockade, such as increased eicosanoid [34] and IL-1 release [35], or alterations in PMN survival. In contrast with known glucocorticoid enhancement of thymocyte apoptosis, however, there is evidence that glucocorticoids inhibit PMN apoptosis [36].
Despite clear effects of endogenous glucocorticoids on PMN migration to the joint in the current study, we have failed to demonstrate an effect on PMN phagocytosis and phagocytosis- or ionophore-induced ROS production. These results are consistent with those of Suzuki et al., who described no alteration in nitroblue tetrazolium-positive PMN counts in one strain of rats following adrenalectomy, despite effects on microvascular adherence [33]. This apparent uncoupling of glucocorticoid effects on PMN migration and activation is also in keeping with the results of studies of the effects of pharmacologic glucocorticoids in animals and in humans [37,38]. In contrast, other studies have reported glucocorticoid modulation of PMN oxidative metabolism [39,40]. The field is complicated by the range of glucocorticoids and doses used in various studies, and by the fact that observation with pharmacologic doses of glucocorticoids frequently does not reflect the effects of endogenous glucocorticoids on immune cell function [21].
In conclusion, the hypothesis that HPA axis dysfunction may lead to failure to attenuate inflammation, and that this dysregulation may, in part, contribute to chronic inflammatory disease is increasingly accepted [41], but no prior studies have examined the effects of endogenous glucocorticoids on synovial PMN recruitment or activation. In the carrageenan arthritis model, we have demonstrated that endogenous glucocorticoids modulate both disease susceptibility and severity. We have further shown that induction of this model is associated with synovial expression of P-selectin, and that endogenous glucocorticoids modulate synovial P-selectin expression. In contrast, PMN phagocytic and oxidative function were not demonstrably influenced by endogenous glucocorticoids in this model. Although it is conceivable that these findings might be restricted to this model of acute arthritis, involvement of both the HPA axis and PMN has been demonstrated in both human RA and other more complex animal models of RA such as adjuvant arthritis. Further studies in suitable systems are required to generalize the current results. The ability of endogenous glucocorticoids to differentially influence components of the inflammatory response and the identification of P-selectin as a target of endogenous glucocorticoid effects have important potential pathologic and therapeutic implications for inflammatory diseases such as RA.
Acknowledgments
This study was supported by the Arthritis Foundation of Australia and the National Health & Medical Research Council, Australia.
References
- 1.Yang YH, Hutchinson P, Leech M, Morand EF. Exacerbation of adjuvant arthritis by adrenalectomy is associated with reduced leukocyte lipocortin 1. J Rheumatol. 1997;24:1758–64. [PubMed] [Google Scholar]
- 2.Mason D, MacPhee I, Antoni F. The role of the neuroendocrine system in determining genetic susceptibility to experimental allergic encephalomyelitis in the rat. J Immunol. 1990;70:1–5. [PMC free article] [PubMed] [Google Scholar]
- 3.Sternberg EM, Hill JM, Chrousos GP, et al. Inflammatory mediator-induced hypothalamic–pituitary–adrenal axis activation is defective in streptococcal cell wall arthritis susceptible Lewis rats. Proc Natl Acad Sci USA. 1989;86:2374–8. doi: 10.1073/pnas.86.7.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harkness JAL, Richter MB, Panayi GS, et al. Circadian variation in disease activity in rheumatoid arthritis. Brit Med J. 1982;284:551–4. doi: 10.1136/bmj.284.6315.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saldanha C, Tougas G, Grace E. Evidence for anti-inflammatory effect of normal circulating plasma cortisol. Clin Exp Rheumatol. 1986;4:365–6. [PubMed] [Google Scholar]
- 6.Saperstein A, Brand H, Audhya T, et al. Interleukin 1 beta mediates stress-induced immunosuppression via corticotropin-releasing factor. Endocrinol. 1992;130:152–8. doi: 10.1210/endo.130.1.1309324. [DOI] [PubMed] [Google Scholar]
- 7.Szabo C, Thiemermann C, Wu C-C, Perretti M, Vane JR. Attenuation of the induction of nitric oxide synthase by endogenous glucocorticoids accounts for endotoxin tolerance in vivo. Proc Natl Acad Sci USA. 1994;91:271–5. doi: 10.1073/pnas.91.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rassnick S, Sved AF, Rabin BS. Locus coeruleus stimulation by corticotropin-releasing hormone suppresses in vitro cellular immune responses. J Neurosci. 1994;14:6033–40. doi: 10.1523/JNEUROSCI.14-10-06033.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jondal M, Okret S, McConkey D. Killing of immature CD4+ CD8+ thymocytes in vivo by anti-CD3 or 5′-(N-ethyl)-carboxamide adenosine is blocked by glucocorticoid receptor antagonist RU-486. Eur J Immunol. 1993;23:1246–50. doi: 10.1002/eji.1830230608. [DOI] [PubMed] [Google Scholar]
- 10.Santos LL, Morand EF, Hutchinson P, Boyce NW, Holdsworth SR. Anti-neutrophil monoclonal antibody therapy inhibits the development of adjuvant arthritis. Clin Exp Immunol. 1997;107:248–54. doi: 10.1111/j.1365-2249.1997.263-ce1154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward PA. Neutrophils and adjuvant arthritis. Clin Exp Immunol. 1997;107:227–9. doi: 10.1111/j.1365-2249.1997.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koch AE, Kunkel SL, Burrows JC, et al. Synovial tissue macrophages as a source of the chemotactic cytokine IL-8. J Immunol. 1991;147:2187–95. [PubMed] [Google Scholar]
- 13.Grober JS, Bowen BL, Ebling H, et al. Monocyte–endothelial adhesion in chronic rheumatoid arthritis. J Clin Invest. 1993;91:2609–12. doi: 10.1172/JCI116500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walter UM, Issekutz AC. The role of E- and P-selectin in neutrophil and monocyte migration in adjuvant-induced arthritis in the rat. Eur J Immunol. 1997;27:1498–505. doi: 10.1002/eji.1830270628. [DOI] [PubMed] [Google Scholar]
- 15.Littler AJ, Buckley CD, Wordsworth P, Collins I, Martinson J, Simmons DL. A distinct profile of six soluble adhesion molecules (ICAM-1, ICAM-3, VCAM-1, E-selectin, L-selectin and P-selectin) in rheumatoid arthritis. Br J Rheumatol. 1997;36:164–9. doi: 10.1093/rheumatology/36.2.164. [DOI] [PubMed] [Google Scholar]
- 16.Hosaka S, Shah MR, Pope RM, Koch AE. Soluble forms of P-selectin and intercellular adhesion molecule-3 in synovial fluids. Clin Immunol Immunopathol. 1996;78:276–82. doi: 10.1006/clin.1996.0039. [DOI] [PubMed] [Google Scholar]
- 17.Youssef PP, Cormack J, Evill CA, et al. Neutrophil trafficking into inflamed joints in patients with rheumatoid arthritis, and the effects of methylprednisolone. Arthritis Rheum. 1996;39:216–25. doi: 10.1002/art.1780390207. [DOI] [PubMed] [Google Scholar]
- 18.Youssef P, Roberts-Thomson P, Ahern M, Smith M. Pulse methylprednisolone in rheumatoid arthritis: effects on peripheral blood and synovial fluid neutrophil surface phenotype. J Rheumatol. 1995;22:2065–71. [PubMed] [Google Scholar]
- 19.van den Brink HR, Van Wijk MJG, Geertzen RGM, Bijlsma JWJ. Influence of corticosteroid pulse therapy on the serum levels of soluble interleukin 2 receptor, interleukin 6 and interleukin 8 in patients with rheumatoid arthritis. J Rheumatol. 1994;21:430–4. [PubMed] [Google Scholar]
- 20.Cronstein BN, Kimmel SC, Levin RI, Martiniuk F, Weissmann G. A mechanism for the antiinflammatory effects of glucocorticoids; the glucocorticoid receptor regulates leukocyte adhesion to endothelial cells and expression of endothelial-leukocyte adhesion molecule 1 and intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1992;86:9991–5. doi: 10.1073/pnas.89.21.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilckens T, de Rijk R. Glucocorticoids and immune function: unknown dimensions and new frontiers. Immunol Today. 1997 doi: 10.1016/s0167-5699(97)01111-0. [DOI] [PubMed] [Google Scholar]
- 22.Bohmer RH, Trinkle LS, Staneck JL. Dose effects of LPS on neutrophils in a whole blood flow cytometric assay of phagocytosis and oxidative burst. Cytometry. 1992;13:525–31. doi: 10.1002/cyto.990130512. [DOI] [PubMed] [Google Scholar]
- 23.Emmendorffer A, Hecht M, Lohmann-Matthes ML, Roesler J. A fast and easy method to determine the production of reactive oxygen intermediates by human and murine phagocytes using dihydrorhodamine 123. J Immunol Methods. 1990;131:269–75. doi: 10.1016/0022-1759(90)90198-5. [DOI] [PubMed] [Google Scholar]
- 24.Tipping PG, Huang XR, Berndt MC, Holdsworth SR. P-selectin directs T lymphocyte-mediated injury in delayed-type hypersensitivity responses: studies in glomerulonephritis and cutaneous delayed-type hypersensitivity. Eur J Immunol. 1996;26:454–60. doi: 10.1002/eji.1830260228. [DOI] [PubMed] [Google Scholar]
- 25.Mc Curdy L, Winn Chatham W, Blackburn WD. Rheumatoid synovial fibroblast adhesion to human articular cartilage. Arthritis Rheum. 1995;38:1694–700. doi: 10.1002/art.1780381123. [DOI] [PubMed] [Google Scholar]
- 26.Leirisalo-Repa M, Paimela L, Koskimies S, Repo H. Functions of polymorphonuclear leukocytes in early rheumatoid arthritis. Inflammation. 1993;17:427–42. doi: 10.1007/BF00916583. [DOI] [PubMed] [Google Scholar]
- 27.Cassatella MA. The production of cytokines by polymorphonuclear neutrophils. Immunol Today. 1995;16:21–26. doi: 10.1016/0167-5699(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 28.Lloyd AR, Oppenheim JJ. Poly's lament: the neglected role of the polymorphonuclear neutrophil in the afferent limb of the immune response. Immunol Today. 1992;13:169–72. doi: 10.1016/0167-5699(92)90121-M. [DOI] [PubMed] [Google Scholar]
- 29.Albert L, Erickson JE, Mason LE, et al. Recombinant human soluble P-selectin glycoprotein ligand-1 decreases adjuvant-induced arthritis in Lewis rats. Arthritis Rheum. 1997;40(Suppl.):S. [Google Scholar]
- 30.Hall J, Morand EF, Medbak S, et al. Abnormal hypothalamo–pituitary–adrenal axis function in human rheumatoid arthritis: effects of nonsteroidal antiinflammatory drugs and water immersion. Arthritis Rheum. 1994;37:1132–7. doi: 10.1002/art.1780370804. [DOI] [PubMed] [Google Scholar]
- 31.Chikanza IC, Petrou P, Kingsley G, Chrousos GP, Panayi GS. Defective hypothalamic response to immune and inflammatory stimuli in patients with rheumatoid arthritis. Arthritis Rheum. 1992;35:1281–8. doi: 10.1002/art.1780351107. [DOI] [PubMed] [Google Scholar]
- 32.Johnson RC, Mayadas TN, Frenette PS, et al. Blood cell dynamics in P selectin-deficient mice. Blood. 1995;86:1106–14. [PubMed] [Google Scholar]
- 33.Suzuki H, Zweifach BW, Forrest MJ, Schmid-Schonbein GW. Modification of leukocyte adhesion in spontaneously hypertensive rats by adrenal corticosteroids. J Leukocyte Biol. 1995;57:20–26. [PubMed] [Google Scholar]
- 34.Vishwanath BS, Frey FJ, Bradbury MJ, Dallman MF, Frey BM. Glucocorticoid deficiency increases phospholipase A2 activity in rats. J Clin Invest. 1993;92:1974–80. doi: 10.1172/JCI116791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perretti M, Becherucci C, Scapigliati G, Parente L. The effect of adrenalectomy on interleukin-1 release in vitro and in vivo. Brit J Pharmacol. 1989;98:1137–42. doi: 10.1111/j.1476-5381.1989.tb12657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox G. Glucocorticoid treatment inhibits apoptosis in human neutrophils: separation of survival and activation outcomes. J Immunol. 1995;154:4719–25. [PubMed] [Google Scholar]
- 37.Llewellyn-Jones CG, Hill SL, Stockley RA. Effect of fluticasone proprionate on neutrophil chemotaxis, superoxide generation, and extracellular proteolytic activity in vivo. Thorax. 1994;49:207–12. doi: 10.1136/thx.49.3.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lomas DA, Chamba IA, Stockley RA. The effect of in vivo and in vitro dexamethasone on human neutrophil function. Agents Actions. 1991;33:279–85. doi: 10.1007/BF01986574. [DOI] [PubMed] [Google Scholar]
- 39.Schleimer RP, Freeland HS, Peters SP, Brown KE, Derse CP. An assessment of the effects of glucocorticoids on degranulation, chemotaxis, binding to vascular endothelium and formation of leukotriene B4 by purified human neutrophils. J Pharm Expr. 1989;250:598–605. [PubMed] [Google Scholar]
- 40.Fuenfer MD, Carr EA, Polk HC. The effect of hydrocortisone on superoxide production by leukocytes. J Surg Res. 1979;27:29–34. doi: 10.1016/0022-4804(79)90106-9. [DOI] [PubMed] [Google Scholar]
- 41.Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocrine Rev. 1984;5:25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]