

Abstract
With a view to determining whether production of Th2 cytokines, IL-4 or IL-10, is responsible for the inability of mice to resolve infection with Mycobacterium tuberculosis, mice with targeted disruption of their IL-4 or IL-10 gene were compared with wild-type mice in terms of their ability to defend against an M. tuberculosis infection initiated via the respiratory route. The results show that mice that are unable to make either IL-4 or IL-10 are no more capable than wild-type mice at defending against tuberculosis (TB). Therefore, the results are inconsistent with the proposition that the inadequacy of Th1-mediated anti-tuberculosis immunity is due to its down-regulation by either of these Th2 cytokines.
Keywords: Th1/Th2, cytokines, anti-mycobacterial immunity
INTRODUCTION
Immunity of mice to infection with Mycobacterium tuberculosis is mediated predominantly by antigen-specific CD4 T cells [1,2], with CD8 T cells also playing a critical role according to some investigators [3], although not according to others [4]. There is evidence [2], in addition, that γδ T cells are involved in anti-tuberculosis immunity. It is generally believed that CD4 cells function to activate the mycobactericidal function of macrophages that need to ingest and kill M. tuberculosis intracellularly. In order for immunity to be expressed by macrophages, interferon-gamma (IFN-γ) needs to be produced by CD4 T cells, as evidenced by the demonstration [5,6] that mice with targeted disruption of their IFN-γ gene are unable to control bacterial multiplication and quickly succumb to infection. Thus, according to the T helper 1/T helper 2 (Th1/Th2) paradigm, T cell-mediated immunity to tuberculosis (TB) is mediated by IFN-γ-secreting, Th1 CD4 cells, as opposed to Th2 CD4 cells which secrete IL-4, IL-10 and other cytokines that are involved in antibody production. Th2 T cells are also believed to function, via the secretion of IL-4 and IL-10, to down-regulate the Th1 response to a variety of microbial pathogens and consequently prevent infection from being resolved [7–10]. It was recently revealed [11,12], in this regard, that during the first 3–4 weeks of an M. tuberculosis infection, mice predominantly generate a Th1 response and produce Th1 cytokines, whereas they also generate a Th2 response and produce Th2 cytokines during the weeks that follow. According to the Th1/Th2 paradigm, this could be interpreted as indicating that an M. tuberculosis-induced Th2 response causes down-regulation of an already ongoing Th1 response that is needed to resolve infection [13]. Hence the reason for the chronicity of M. tuberculosis infection. The presence to a greater or lesser degree of Th2 cytokines in the peripheral circulation of humans with active TB has likewise been used to explain why the disease is more severe in some humans than in others [14]. The same argument has been used to explain the inadequacy of the immune response to leprosy [15]. Moreover, because IL-10 can be produced by a variety of cells besides Th2 CD4 cells, and can inhibit macrophage microbicidal activity directly [16–18], its suppressive action on macrophage function has been offered as an explanation of why these cells fail to eliminate M. tuberculosis from sites of infection [19].
The purpose of the study described here was to investigate whether mice that are incapable of producing either IL-4 or IL-10, because of targeted disruption of their IL-4 or IL-10 gene, respectively, are more capable than wild-type mice at defending themselves against M. tuberculosis infection.
MATERIALS AND METHODS
Mice
IL-4, IL-10 and IFN-γ gene-disrupted mice, as well as wild-type mice, were on a B6129 (C57Bl/6 × 129/SvJ) background. They were obtained from the Jackson Laboratories (Bar Harbor, ME), and employed in experiments at 8–10 weeks of age. All mice were housed under specific pathogen-free conditions.
Bacteria
Mycobacterium tuberculosis, strain H37Rv, was obtained from the Trudeau Institute Mycobacterial Culture Collection, and prepared for infecting mice via the respiratory route in a Middlebrook Airborne Infection Apparatus, as described previously [20]. The organism was enumerated in infected organs by plating serial 10-fold dilutions of whole organ homogenates on nutrient agar, and counting colonies after 2–3 weeks incubation at 37°C, as described previously [21].
RESULTS
The results in Figs.1 and 2 show that mice devoid of a capacity to generate either IL-4 or IL-10 were the same as wild-type mice in terms of their ability to defend against M. tuberculosis infection initiated via the respiratory route with about 102 bacilli. In all groups of mice, infection in the lungs progressed for about 20 days before being brought under control and caused to plateau for the next 70 days. In the liver and spleen M. tuberculosis was not present in detectable numbers until about day 20 of infection in all cases. After this time infection in these organs either plateaued or very slowly progressed.
Fig. 1.
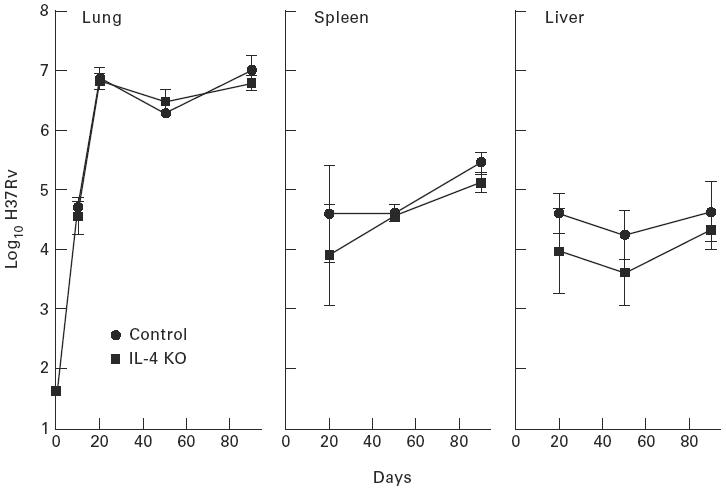
Growth of Mycobacterium tuberculosis in the lungs, livers and spleens of wild-type and IL-4 gene-disrupted mice infected with 101.6 bacilli by aerosol. Inability to make IL-4 made no significant difference to the ability of mice to control infection. Means ± s.d. of five mice per group.
Fig. 2.
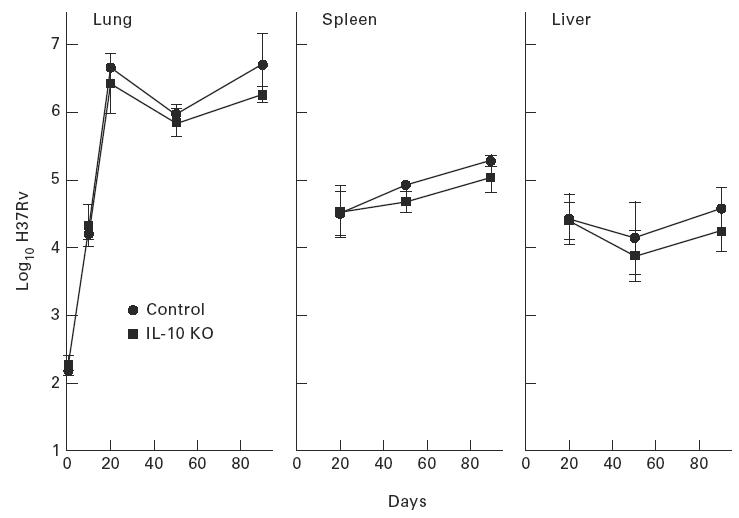
Wild-type and IL-10 gene-disrupted mice are compared in terms of their ability to defend against Mycobacterium tuberculosis infection initiated with 102.2 bacilli given by aerosol. Both types of mice were equally capable of defending against infection, as measured by growth of the pathogen in their lungs, livers and spleens. Means ± s.d. of five mice per group.
Evidence that stabilization of infection in the lungs of wild-type mice after day 20 of infection, as shown in Fig.1, is under the control of a Th1 immune response is supplied in Fig.3, which shows the course of infection in mice with a functionally disrupted IFN-γ gene. It can be seen that in the absence of a capacity to make IFN-γ mice were highly susceptible to infection, in that the pathogen grew progressively in all organs beyond day 20. These mice began dying on day 40.
Fig. 3.
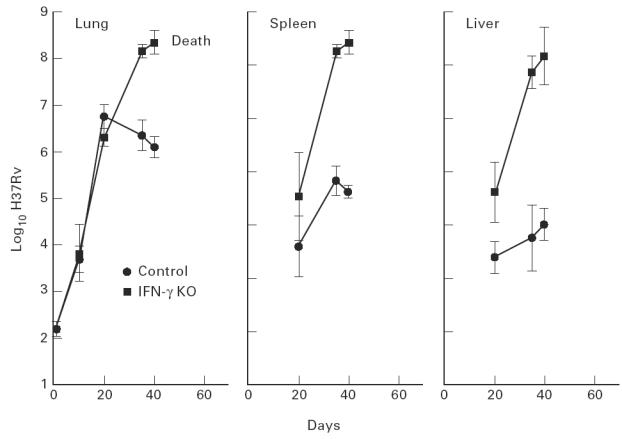
IFN-γ gene-disrupted mice, in contrast to wild-type mice, were incapable of controlling Mycobacterium tuberculosis growth in their lungs, livers and spleens after day 20 of an infection initiated with 102 bacilli by aerosol. The former mice began dying on about day 40 of infection. Means ± s.d. of five mice per group.
DISCUSSION
This study deals with the question of whether failure of Th1 CD4 T cell-mediated immunity to completely resolve infection with virulent M. tuberculosis in mice is due to the negative immunoregulatory influence of IL-4 or IL-10. As a way of illustrating that infection with this pathogen induces a partially protective state of Th1 CD4 T cell-mediated immunity, it is shown, in agreement with the finding of others [5,6], that unlike wild-type mice that acquire the capacity to stabilize the level of infection from about day 20 on, mice with a disrupted IFN-γ gene are unable to control the progression of infection in their lungs, livers and spleens, and consequently begin dying on about day 40. Because the consequence of depleting mice of CD4 T cells [21] is similar to that of removing the capacity to make IFN-γ, the results are in keeping with the generally held view [1,2] that immunity responsible for stabilizing infection is mediated predominantly by Th1 CD4 T cells. An essential role for IL-12 in initiating this Th1 response was recently demonstrated using mice with targeted disruption of their IL-12 p40 gene [22].
The results presented here demonstrate that absence of an ability to produce IL-4 or IL-10 has no effect on the course of infection with M. tuberculosis. Therefore, failure of Th1 CD4-mediated immunity to completely resolve infection need not depend on the negative immunoregulative function of either of these key Th2 cytokines. The situation with M. tuberculosis infection therefore is different from that with leishmaniasis, where neutralization of IL-4 alone with anti-IL-4 antibody enables otherwise susceptible mice to resolve infection [23]. It should be kept in mind, moreover, that it has been shown [24] that mice with a disrupted IL-4 gene are severely deficient in a capacity to generate a Th2 response and to produce significant quantities of other Th2 cytokines. This is in keeping with the need for IL-4 to engage the IL-4 receptor on the surface of CD4 cells, in order for Th2 cytokine transcription to be initiated via activation of Stat6 [25]. This would seem to be the reason why IL-4 is used exclusively in vitro to cause polarization of activated CD4 T cells to a Th2 phenotype. It is likely therefore that in the absence of IL-4, other Th2 cytokines are not produced by CD4 cells in response to M. tuberculosis infection. This would still leave IL-10 produced by macrophages and other cells to possibly suppress initiation of a Th1 response [26], or to directly suppress the function of infected macrophages and prevent them from expressing microbicidal activity [10,19]. However, the results presented here with IL-10 knockout mice show that this down-regulatory activity of IL-10 cannot by itself explain the chronicity of mouse TB. In the absence of any direct evidence that anti-tuberculosis immunity is, in fact, suppressed in vivo, it may be more profitable to consider other reasons for the chronicity of TB. For example, the ability of M. tuberculosis to become dormant under the pressure of host immunity [27] could explain why it can persist in the tissues in the face of acquired immunity.
Lastly, it has been argued in recent publications [20,21,28] that mouse TB is a relevant model with which to study important aspects of the human disease. The argument is based on the knowledge that, in mice [20,28], as in most humans [29], TB is a disease confined to the lungs, even though M. tuberculosis disseminates via blood and implants in other organs. Because in immunocompromised mice [21] and in immunocompromised humans [30], infection can become systemic and cause pathology in multiple organs, it follows that in immunocompetent mice and humans infection-induced pathology is confined to the lungs, because immunity is expressed efficiently in all organs except the lungs. However, unpublished results of experiments in this laboratory show that mouse TB only resembles human TB if infection is initiated with a small enough number of bacilli to ensure long enough survival for chronic lung pathology of the type seen in humans to develop. Hence the reason in the present study for infecting mice with about 102 colony-forming units of M. tuberculosis via the respiratory route.
Acknowledgments
The author would like to thank Ron LaCourse and Lynn Ryan for expert technical support. This research was supported by grants HL-51960, AI-40071 and AI-37844 from the United States Public Health Service.
REFERENCES
- 1.Orme IA, Andersen P, Boom WH. T cell response to Mycobacterium tuberculosis. J Infect Dis. 1993;167:1481–9. doi: 10.1093/infdis/167.6.1481. [DOI] [PubMed] [Google Scholar]
- 2.Boom WH. The role of T cell subsets in Mycobacterium tuberculosis infection. Infect Agents Dis. 1996;5:73–81. [PubMed] [Google Scholar]
- 3.Flynn JL, Goldstein MA, Treibold KJ, Koller B, Bloom BR. Major histocompatibility complex class 1-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc Natl Acad Sci USA. 1992;89:12013–7. doi: 10.1073/pnas.89.24.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leveton C, Barnass S, Champion B, Lucas S, De Sousa B, Nicol M, Banerjee D, Rook G. T-cell-mediated protection of mice against virulent Mycobacterium tuberculosis. Infect Immun. 1989;57:390–5. doi: 10.1128/iai.57.2.390-395.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russel DG, Orme IM. Disseminated tuberculosis in gamma gene-disrupted mice. J Exp Med. 1993;178:2243–7. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flynn JL, Chan J, Tiebold KJ, Dalton DK, Stewert TA, Bloom BR. An essential role for interferon gamma in resistance in Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–54. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosmann TR, Moore KW. The role of IL-10 in cross regulation of Th1 and Th2 response. Immunol Today. 1991;12:A49–A53. doi: 10.1016/S0167-5699(05)80015-5. [DOI] [PubMed] [Google Scholar]
- 8.Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today. 1996;17:138–46. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 9.Abbas AK, Murphy KM, Sher AL. Functional diversity of helper T cells. Nature. 1996;383:787–93. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 10.Dipiro JT. Cytokine networks with infection: mycobacterial infection, leishmaniasis, human immunodeficiency virus infection, and sepsis. Pharmacother. 1997;17:205–23. [PubMed] [Google Scholar]
- 11.Orme IM, Roberts AD, Griffin JP, Abrams J. Cytokine secretion by CD4 lymphocytes in response to Mycobacterium tuberculosis infection. J Immunol. 1995;151:518–24. [PubMed] [Google Scholar]
- 12.Hernandez-Pando R, Orozcoe H, Sampieri A, Pavon L, Velasquillo C, Larriva-Sahd J, Alcocer JM, Madrid MV. Correlation between the kinetics of Th1/Th2 cells and pathology in a murine model of experimental pulmonary tuberculosis. Immunology. 1996;89:26–33. [PMC free article] [PubMed] [Google Scholar]
- 13.Huygen K, Abramowicz D, Vanenbussche P, et al. Spleen cell cytokine secretion in Mycobacterium bovis BCG-infected mice. Infect Immun. 1992;60:2880–6. doi: 10.1128/iai.60.7.2880-2886.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dlugovtzky D, Torres-Morales A, Rateni L, Farron MA, Largach C, Molteni O, Bottasso O. Circulating profile of Th1/Th2 cytokines in tuberculosis patients with different degrees of pulmonary involvement. FEMS Immunobiol Med Microbiol. 1997;18:203–7. doi: 10.1111/j.1574-695X.1997.tb01046.x. [DOI] [PubMed] [Google Scholar]
- 15.Sieling PA, Modlin RL. Cytokine patterns at the site of mycobacterial infection. Immunobiol. 1994;191:378–87. doi: 10.1016/S0171-2985(11)80443-2. [DOI] [PubMed] [Google Scholar]
- 16.Bogdan C, Vodovotz Y, Nathan C. Macrophage deactivation by interleukin-10. J Exp Med. 1991;174:1549–55. doi: 10.1084/jem.174.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore KW, O'Garra A, de Waal Malefyt R, Vieira P, Mosmann TR. Interleukin-10. Ann Rev Immunol. 1993;11:165–90. doi: 10.1146/annurev.iy.11.040193.001121. [DOI] [PubMed] [Google Scholar]
- 18.Rennick DA, Fort MF, Davidson NJ. Studies with IL-10−/– mice: an overview. J Leukocyte Biol. 1997;61:389–97. doi: 10.1002/jlb.61.4.389. [DOI] [PubMed] [Google Scholar]
- 19.Murry PJ, Wang L, Onufryk C, Tepper RI, Young RA. T cell-derived IL-10 antagonizes macrophage function in mycobacterial infection. J Immunol. 1997;158:315–21. [PubMed] [Google Scholar]
- 20.Medina E, North RJ. Evidence inconsistent with a role for the Bcg gene (Nramp1) in resistance of mice to infection with virulent Mycobacterium tuberculosis. J Exp Med. 1996;183:1045–51. doi: 10.1084/jem.183.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunn PL, North RJ. Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology and cause mortality in mice. Infect Immun. 1995;63:3428–37. doi: 10.1128/iai.63.9.3428-3437.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooper AM, Magram J, Farrante J, Orme IM. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with Mycobacterium tuberculosis. J Exp Med. 1997;186:39–45. doi: 10.1084/jem.186.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coffman RL, Varkila K, Scott P, Chatelain R. Role of cytokines in the differentiation of T-cell subsets in vivo. Immunol Rev. 1991;123:189–207. doi: 10.1111/j.1600-065x.1991.tb00611.x. [DOI] [PubMed] [Google Scholar]
- 24.Kopf M, Gros GL, Bachman M, Lamers MC, Bluethmann H, Kohler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362:245–8. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 25.Takeda K, Tanaka T, Shi W, et al. Essential role of Stat6 in IL-4 signaling. Nature. 1996;380:627–30. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh C, Heimberger AB, Gold JS, O'Garra A, Murphy KM. Differential regulation of helper phenotype development by interleukins 4 and 10 in αβ T-cell-receptor transgenic system. Proc Natl Acad Sci USA. 1992;89:6065–9. doi: 10.1073/pnas.89.13.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheeler PR, Ratlege C. Metabolism of Mycobacterium tuberculosis. In: Bloom BR, editor. Tuberculosis: pathogenesis, protection and control. Washington DC: ASM Press; 1994. pp. 353–85. [Google Scholar]
- 28.Dunn PL, North RJ. Persistent infection with virulent but not avirulent Mycobacterium tuberculosis in the lungs of mice causes progressive pathology. Med Microbiol. 1996;45:103–9. doi: 10.1099/00222615-45-2-103. [DOI] [PubMed] [Google Scholar]
- 29.Farer LS, Lowell AM, Meador MP. Extrapulmonary tuberculosis in the United States. Am J Epidemiol. 1979;109:205–17. doi: 10.1093/oxfordjournals.aje.a112675. [DOI] [PubMed] [Google Scholar]
- 30.Barns PF, Block AB, Davidson PT, Snider DE. Tuberculosis in patients with human immunodeficiency virus infection. N Engl J Med. 1991;324:1644–50. doi: 10.1056/NEJM199106063242307. [DOI] [PubMed] [Google Scholar]