

Abstract
Goodpasture's disease, an autoimmune disorder causing severe glomerulonephritis and pulmonary haemorrhage, is characterized by antibodies to the glomerular basement membrane (GBM). The principal target antigen has been identified as the carboxyl terminal non-collagenous (NC1) domain of the α3-chain of type IV collagen. Anti-GBM antibodies appear to recognize one major epitope that is common to all patients, and is largely conformational. We have analysed antibody binding to recombinant α(IV)NC1 domains using a construct and expression system shown to produce correctly folded antigen that is strongly recognized by autoantibodies. In this system, as with the native antigen, α3(IV)NC1 was bound strongly by antibodies from all patients, whereas the closely related α1(IV) and α5(IV)NC1 domains, similarly expressed, showed no such binding. A series of chimeric NC1 domains, between human α3(IV) and α1(IV), and between human and rat α3(IV), were expressed as recombinant molecules, and were recognized by autoantibodies to varying degrees. Strong binding required the presence of human α3(IV) sequence in the amino terminal region of both sets of chimeric molecules. This work strongly suggests that the amino terminal of α3(IV)NC1 is critical for antibody recognition, whereas the carboxyl terminal end of α3(IV)NC1 has a less important role.
Keywords: type IV collagen, glomerular basement membrane, Goodpasture antigen, autoantibody binding, B cell epitope
INTRODUCTION
Goodpasture's disease is a rare autoimmune disorder, characterized by autoantibodies to the glomerular basement membrane (GBM), and often causing rapidly progressive glomerulonephritis and lung haemorrhage [1–3]. The pathogenicity of the autoantibodies has been shown by transfer experiments in primates, and is supported by clinical observations [4]. The aetiology of Goodpasture's disease is understood better than most forms of nephritis, making it a particularly good model in which to study mechanisms of autoimmunity.
The specific target of the autoantibodies, the Goodpasture antigen, has been localized to the non-collagenous (NC1) domain of the α3-chain of type IV collagen chain [5–8]. This is one of six related α(IV) chains, the ubiquitous α1(IV) and α2(IV) chains and the tissue-specific α3(IV)-α6(IV) chains [9]. A typical type IV collagen chain is ≈ 1700 residues in length, with a relatively short 7S domain at the amino end, a long collagenous region characterized by Gly-Xaa-Yaa repeats with short non-collagenous interruptions, and a non-collagenous (NC1) domain of 230 residues at the carboxyl terminus. Type IV collagen NC1 domains contain a highly conserved pattern of 12 cysteine residues, which is common to all known type IV collagen chains. These are believed to form six intramolecular disulphide bonds essential to the overall tertiary structure of the molecule [10]. Binding of Goodpasture antibodies to the autoantigen is conformationally dependent, as reduction of the disulphide bridges and denaturation results in abrogation of antibody binding [11,12].
The six α(IV)NC1 domains can be divided into two subgroups; the ‘α1-like’ group in which α3(IV)NC1 and α5(IV)NC1 show a homology of 74% and 82%, respectively, with α1(IV)NC1; and the ‘α2-like’ group in which α4(IV)NC1 and α6(IV)NC1 show a homology of 73% and 79%, respectively, with α2(IV)NC1 [13–16]. There is considerably less homology between the two subgroups, of between 54% and 64%. The α1(IV) and α2(IV) chains are expressed ubiquitously in basement membranes and their genes are arranged head-to-head on chromosome 13 with a common promoter [17]. The α3(IV) and α4(IV) chain genes share a similar arrangement on chromosome 2 [18–20], but are expressed only in certain specialized basement membranes, including those of the glomerular and the alveolar basement membranes [21,22]. Despite the high degree of homology between the α(IV)NC1 domains, only α3(IV)NC1 binds Goodpasture autoantibodies consistently, although some sera appear to bind other α(IV)NC1 domains weakly [23,24].
In this study, we investigated the epitope specificity of Goodpasture autoantibodies in the autoimmune response. B cell-mediated antigen presentation could be an important mechanism for both initiating [25] and perpetuating autoimmunity. Antigen uptake mediated by the specific immunoglobulin receptors on the surface of B cells allows them to be two orders of magnitude more efficient at stimulating T cells than other antigen-presenting cells (APC) exposed to the same concentration of antigen internalized non-specifically [26]. Furthermore, the specificity of the autoantibody may favour the presentation of particular peptides by APC (possibly peptides that would never reach ‘significant’ levels in normal circumstances), presumably by altering the proteolytic fate of parts, or the whole molecule [27]. These mechanisms are therefore of great potential importance in autoimmune diseases that are associated with autoantibodies.
Previous studies have investigated the Goodpasture epitope by studying synthetic peptides based on short linear sequences within the protein. Peptide inhibition studies suggested that the Goodpasture epitope was localized in the carboxyl region of the α3(IV)NC1 domain [28]. However, other authors were unable to demonstrate antibody binding to peptides derived from this region [29], but found consistent binding to several other peptides throughout the molecule. Research involving the expression of splicing variants of α3(IV)NC1 showed that the carboxyl region was not essential for antibody binding [30]. Our own studies comparing the sequence of the antigen in different species have suggested that the amino region was important for antibody recognition [31]. This work allowed us to identify regions of potential importance and eliminate others, but at best can only be a broad guideline.
Conclusions about conformational epitopes that are based on studies with synthetic peptides and malfolded polypeptides must necessarily be tentative, therefore we sought an alternative approach. In this study we adapted a mammalian expression system to produce a recombinant mini-collagen chain form of human α3(IV)NC1, previously shown to be expressed in insect cells [32], which has strong reactivity with patients' sera. We also expressed the closely related human α1(IV)NC1, human α5(IV)NC1, and rat α3(IV)NC1. We went on to generate chimeric molecules, involving the substitution of corresponding segments between homologous NC1 domains. These molecules are likely to retain their three-dimensional structure because of high sequence homologies between the different α(IV)NC1 chains and a highly conserved pattern of 12 cysteine residues which form disulphide bonds crucial to the tertiary structure of all type IV collagen chains. That the molecules do fold correctly has been confirmed by the success of this study. Human α1(IV)NC1 and rat α3(IV)NC1 have 74% and 91% amino acid homology with human α3(IV)NC1, respectively, and are therefore ideal molecules for homologous substitution. In this study we have expressed six chimeras of recombinant human α3(IV)/α1(IV)NC1 and of human/rat α3(IV)NC1, and used them to map the binding of autoantibodies to the Goodpasture antigen. This work defines the B cell epitope(s) of the intact molecule more clearly.
PATIENTS AND METHODS
Patients
Sera were taken from six patients diagnosed as having Goodpasture's disease, with linear antibody deposition on direct immunofluorescence of renal biopsies. The patients were generally representative of the Hammersmith Hospital series; three had pulmonary haemorrhage and nephritis and three had isolated anti-GBM nephritis. Circulating anti-GBM antibodies, from each patient, were detected by ELISA [33] and Western blotting against collagenase-solubilized human GBM [34].
Antibodies
Sera from the Goodpasture patients, stored at −20°C, were diluted 1:30 to detect native and recombinant antigen on nitrocellulose blots. The murine MoAb P1 has been described previously [35]. The murine MoAb W17 was kindly supplied by Dr J. Wieslander [36] (University Hospital, Lund, Sweden).
Preparation of collagenase-solubilized GBM
Glomeruli were isolated from fresh human kidney cortex by differential sieving. GBM (the native antigen) was extracted by sonication and subjected to digestion with collagenase as previously described [19]. Rat GBM was prepared by similar methods as previously described [37].
Construction of type IV mini-collagen chain gene and cloning into mammalian expression vector
All DNA manipulations were carried out using standard procedures [38]. A cDNA of the leader peptide, NH2 terminus and 7S domain of the human α1(IV) chain (a 530-bp fragment), was joined in-frame to the NC1 domain of the human α3(IV) chain, effectively creating a mini-collagen chain gene [32]. This composite cDNA contained a Sal1 site at the 5′ end, a BamH1 site at the junction of α1(IV)/α3(IV), and Xho1, Kpn1 sites at the 3′ end. The Sal1-Xho1 fragment, encoding IL-2, of the plasmid pBC12/CMV/IL-2 [39] was removed by restriction enzyme digestion and gel purification. The Sal1-Xho1 fragment of p530.α3(IV)NC1, encoding the α1(IV)/α3(IV) hybrid mini-collagen chain, was gel- purified and cloned into the Sal1/Xho1-cut pBC12/CMV/IL-2. The resultant plasmid was designated as pCMV 530-α3(IV)NC1.
Cloning of human α1(IV)NC1, human α5(IV)NC1 and rat α3(IV)NC1 cDNA fragments as mini-collagen chain genes
The human α3(IV)NC1 domain of the mini-collagen chain gene was sequentially replaced by the NC1 domains of human α1(IV), human α5(IV) and rat α3(IV). The BamH1/HindIII fragment of the plasmid pHT-21 [40], encoding the human α1(IV)NC1, was subcloned into pGEM, and amplified with the oligonucleotide primers 5′ TTGCCAGGATCCATGGGGCCT 3′ and 5′ TTGGTACCTCGAGGCTTCATTA 3′ to introduce XhoI and KpnI sites in the 3′ non-coding region. The BamHI/XhoI α1(IV)NC1 cDNA fragment was then used to replace the α3(IV)NC1 cDNA fragment in the expression vector described above. The α5(IV)NC1 cDNA was amplified from reverse- transcribed RNA from human renal cortex using the primers 5′ CCATGGATGGTCCCCCT 3′ and 5′ CTCGAGACACTGCATCCTAG 3′, and cloned into pUC12 cut with SmaI. The NcoI/XhoI fragment was subcloned into the same sites of an α3(IV)NC1 cDNA into which BamHI and NcoI sites had been introduced in the collagenous region, and XhoI and KpnI sites into the 3′ non-coding region [32]. The BamH1/XhoI fragment of this plasmid was then used to replace the α3(IV)NC1 cDNA fragment in the expression vector described above. The complete rat α3(IV)NC1 sequence was obtained from a rat kidney cDNA library (Clontech Labs, Basingstoke, UK) and was used to replace the α3(IV)NC1 cDNA fragment in the expression vector described above [41]. Polymerase chain reaction (PCR) amplification was performed using Taq (Perkin Elmer, Branchburg, USA) or Vent (New England Biolabs, Hitchin, UK) polymerases and 30 cycles of the following pattern in a Biometra Trio-thermoblock: 94°C for 1 min, 55°C for 1 min, 72°C for 2 min; followed by a final 10-min step at 72°C.
Construction of human α3(IV)/α1(IV)NC1 chimeras
The α3(IV)NC1 cDNA was divided into three regions based on the restriction enzyme sites PvuII and EcoR1. These regions were designated as amino, central and carboxyl. The human α1(IV)NC1 cDNA sequence has a PvuII site at the position precisely corresponding to that of human α3(IV)NC1. An EcoR1 restriction site, corresponding to that in the α3(IV)NC1 cDNA, was introduced into the α1(IV)NC1 cDNA by site-directed mutagenesis (Clontech), using the primer 5′ TGCCTGGAGGAATTCAGAAGTGCG 3′.
A modified vector was constructed by removing the PvuII fragment of pUC19 and dephosphorylating its blunt-ended arms. This process removed the polylinker from the pUC19 vector without affecting its ability to be used as a cloning vector. The BamH1/Kpn1 fragments of α3(IV)NC1 and α1(IV)NC1 were gel-purified, made blunt-ended using T4 DNA polymerase and nucleotide tri-phosphates (NTP), phosphorylated and cloned into the specially modified pUC19 vector, recreating the BamH1 site. The common regions of α3(IV)NC1 and α1(IV)NC1 were exchanged to create six chimeric molecules (Fig. 1). The chimaeric α3(IV)/α1(IV)NC1 BamH1/Xho1 fragments were then subcloned in BamH1/Xho1-cut pCMV 530-α3(IV)NC1 (pCMV 530), and expressed in COS-7 cells. All constructs were verified by unique restriction enzyme digestion
Fig. 1.
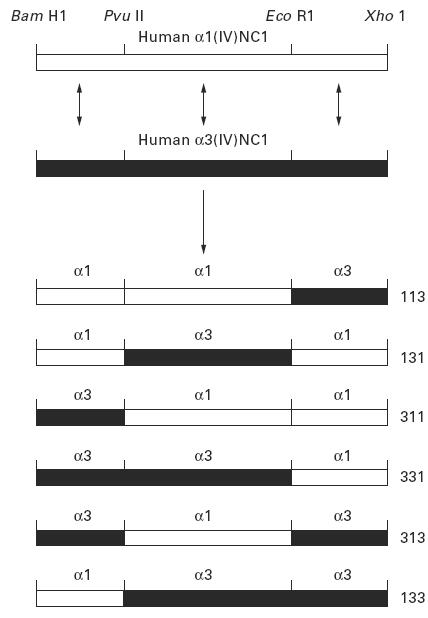
Schematic diagram illustrating the derivation of chimeric human α3(IV)/α1(IV)NC1 domains. Exchange of corresponding regions of α3(IV)NC1 and α1(IV)NC1 domains to create six chimeric α3(IV)/α1(IV)NC1 domains. The constructs are described according to the origins of their amino, central and carboxyl regions, i.e. α331 was derived from the amino and central regions of α3(IV)NC1 and the carboxyl region of α1(IV)NC1.
Construction of human/rat α3(IV)NC1 chimeras
An identical cloning strategy was adopted to construct chimeras of human α3(IV)NC1 and rat α3(IV)NC1 domains using their BamH1/Xho1 fragments. The rat α3(IV)NC1 cDNA has PvuII and EcoR1 sites at the corresponding positions to those of human α3(IV)NC1. The BamH1/Kpn1 fragments of human and rat α3(IV)NC1 were gel-purified, made blunt-ended using T4 DNA polymerase and NTP, phosphorylated and cloned into the specially modified pUC19 vector, recreating the BamH1 site. The common regions of human and rat α3(IV)NC1 were exchanged to create six chimeric molecules. The chimeric human/rat α3(IV)NC1 BamH1/Xho1 fragments were then subcloned into pCMV-530 vector and expressed in COS-7 cells.
Expression of recombinant α(IV)NC1 domains
COS-7 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Paisley, UK) supplemented with 5% fetal calf serum (FCS). Cells were seeded at a density of 2–2.5 × 106/75-cm2 flask overnight, after which a subconfluent layer of cells was transfected with 5 μg of DNA using a DEAE–Dextran-mediated procedure [42]. The plasmid DNA was mixed with 1.9 ml of PBS and 100 μl of DEAE–Dextran (10 mg/ml). COS cells were rinsed with warm PBS (calcium- and magnesium-free), aspirated and the 2 ml transfection cocktail added. Flasks were incubated at 37°C for 30 min, with occasional gentle shaking to prevent drying. Culture medium (8 ml), supplemented with 100 μm of chloroquine, was added to each flask and incubation continued for 2.5 h, after which supernatants were aspirated and cells were treated with 7.5 ml of culture media containing 10% DMSO for 2.5 min. This was replaced by DMEM plus 5% FCS. Transfected cells were incubated for 72 h and supernatants were collected and stored at −20°C. The human and rat α3(IV)NC1 mini-collagen chains, along with the related α1(IV)NC1 and α5(IV)NC1 chains, were expressed in the same manner.
SDS–PAGE and Western blotting
COS supernatants were analysed by electrophoresis on 10% or 12.5% polyacrylamide gels containing SDS [43]. Gels were electroblotted onto nitrocellulose and membranes blocked with PBS/0.5% Tween for 1–2 h. Recombinant protein was detected using a 1:30 dilution of patients' serum (primary antibody) or MoAb in conjunction with alkaline phosphatase-conjugated anti-human or anti-mouse IgG (Sigma-Aldrich, Poole, UK). Colour was developed with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate (Sigma).
Cation exchange purification of recombinant protein
Cation exchange purification of recombinant protein was achieved using a bifunctional cation exchange resin, Bakerbond carboxysulphon. A 100 × 5 mm PEEK column was packed with carboxysulphon, and equilibrated in 0.05 m Tris–HCl buffer pH 7.3. Culture supernatant from transfected COS-7 cells was adjusted to pH 7.3 with 1 m acetic acid. Supernatant (20 ml) was then loaded onto the column at a rate of 1 ml/min and the column was washed for 20 min with Tris buffer. Cationic material bound to the column was eluted using a linear gradient of 0–100% of 0.5 m NaCl in 0.05 m Tris–HCl pH 7.3, over 30 min, and 1-ml fractions were collected. Loading of sample and eluting of fractions were carried out using a Waters 626 high pressure liquid chromatograph (HPLC) system. Fractions containing antigenic material were identified by SDS–PAGE and Western blot analysis. These were pooled, concentrated and desalinated on Centricon C30 (Amicon, Stonehouse, UK) columns and resuspended in 0.5 ml of TE buffer.
ELISA for detection of recombinant α3(IV)NC1
An ELISA for the detection of human or other mammalian GBM, and recombinant α3(IV)NC1, using anti-GBM antibodies, was developed for use in 96-well microtitre plates [44]. Collagenase-solubilized human GBM was coated at a concentration of 40 μg/ml in guanidine thiocynate as a positive control. Recombinant protein was diluted in 50 mm sodium carbonate buffer pH 9.8, and 100 μl were used to coat each well in duplicate. After overnight incubation at 4°C, the plate was washed three times in PBS–Tween (PBS/Tw) and then blocked for 1 h in 100 μl of blocking buffer containing PBS/Tw/1% bovine serum albumin (BSA). Primary antibody, in the form of sera from Goodpasture patients, was diluted in blocking buffer and 100 μl were incubated in each well at 37°C for 1 h. The plate was again washed three times, and 100 μl of 1:1000 dilution of secondary antibody, alkaline phosphatase-conjugated goat anti-IgG, in blocking buffer, were added to each well and incubated for 1 h at 37°C. The plate was washed three times, and developed with phosphatase substrate tablets; one tablet was dissolved in 5 ml carbonate buffer containing 5 mM MgCl2 and 100 μl of this were added to each well. Colour was allowed to develop and the optical density (OD) was read at 405 nm using an Anthos htII ELISA plate reader.
35S-radiolabelling of recombinant protein
Radiolabelling of recombinant protein was carried out in order to compare the protein expression levels of COS-7 cells that were transfected with different chimeric NC1 domain constructs. At 60–72 h after transfection, cell cultures were washed twice in cysteine- and methionine-free DMEM media (−FCS). Cells were then incubated in this medium for 20 min. This depleted the cells of their intracellular pools of sulphur-containing amino acids. The medium was removed by aspiration and 2 ml of fresh warm medium, containing 200 μCi of Promix (35S-labelled methionine and cysteine; Amersham Life Science, Aylesbury, UK) were added to each 75-mm2 flask. Cells were incubated at 37°C for 6 h, after which supernatants were collected and stored at −20°C. The supernatants were run on a 12.5% SDS–PAGE gel. The gels was then fixed in 10 volumes of glacial acetic acid:methanol:water in a ratio of 10:20:70 for 5 min. After a brief wash in distilled water, the gel was placed on saran wrap and a piece of 3MM blotting paper was placed over it and inverted. The gel was dried at 50°C for 90 min and then exposed to autoradiograph film for 48 h.
RESULTS
Characterization of recombinant human α3(IV)NC1 and related chains
COS-7 cells were transfected with 10 μg of plasmid DNA using standard DEAE–dextran techniques. Supernatants were collected after 72 h and examined by SDS–PAGE and Western blotting. Recombinant α3(IV)NC1 was detected in COS supernatants by patients' sera at the predicted molecular size of 41 kD. All six sera studied bound strongly to α3(IV)NC1. Recombinant α1(IV)NC1 was not recognized by patients' sera, while α5(IV)NC1 was weakly recognized by only one patient's serum (Fig. 2). Recombinant α3(IV)NC1 was also recognized by two MoAbs, P1 (data not shown) and W17 (Fig. 5). These two MoAbs have been raised to human and bovine GBM, respectively, and shown to bind to native human α3(IV)NC1 [21,35,36,45]. Recombinant α1(IV)NC1 and α5(IV)NC1 were not recognized by P1 or W17. Recombinant rat α3(IV)NC1 was detected, at the predicted molecular size of 42 kD, by human autoantibodies, but binding was considerably weaker than that to human α3(IV)NC1 (Fig. 3).
Fig. 2.
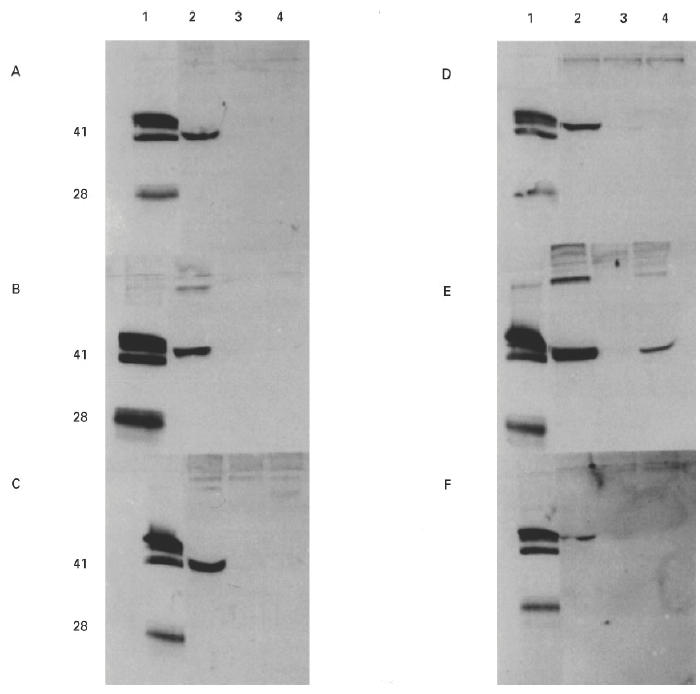
Western blots showing binding of autoantibodies from six patients to recombinant α(IV)NC1 domains. COS supernatants from transfected cells were separated on 12.5% SDS–PAGE gels and electroblotted to nitrocellulose. Recombinant protein was detected with human autoantibodies from Goodpasture patients A–F. Lane 1 contained collagenase-solubilized (cs) human glomerular basement membrane (GBM) (20 μg) as a positive control. The recombinant protein in the other lanes was: lane 2, human α3(IV)NC1; lane 3, human α1(IV)NC1; lane 4, human α5(IV)NC1.
Fig. 5.
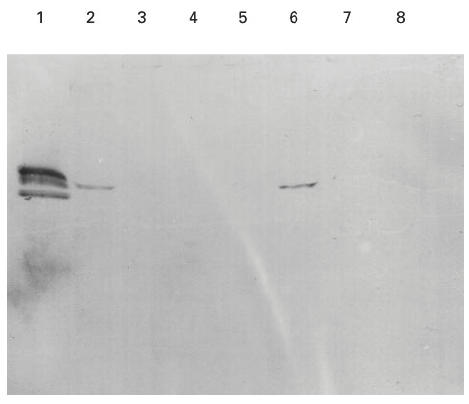
Western blot showing binding of MoAb 17 to chimeric α3/α1(IV)NC1 domains. COS supernatants from transfected cells were separated on 12.5% SDS–PAGE gels and electroblotted to nitrocellulose. Recombinant chimeric human α3/α1(IV)NC1 domains were detected with 1:100 dilution of MoAb W17. Lane 1 contained collagenase-solubilized (cs) human glomerular basement membrane (GBM) as a positive control. The recombinant protein in the other lanes was: lane 2, human α3(IV)NC1 (α333); lane 3, α113; lane 4, α131; lane 5, α311; lane 6, α331; lane 7, α313; lane 8, α133.
Fig. 3.
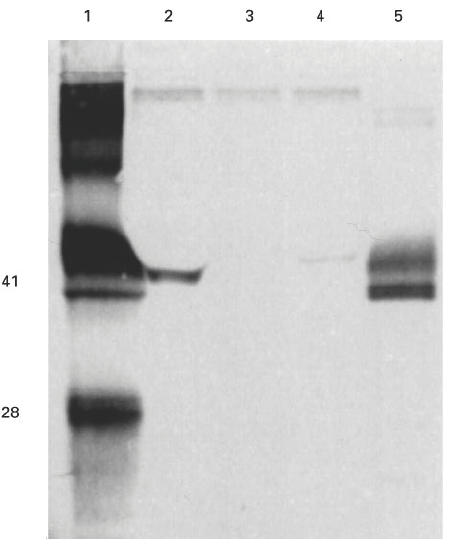
Western blot showing binding of human autoantibodies to recombinant human and rat α(IV)NC1 domains. COS supernatants from transfected cells were separated on 12.5% SDS–PAGE gels and electroblotted to nitrocellulose. Recombinant protein was detected with human autoantibodies from Goodpasture patients. Lanes 1 and 5 contained collagenase-solubilized (cs) human and rat glomerular basement membrane (GBM), respectively, as positive controls. The recombinant protein in the other lanes was: lane 2, human α3(IV)NC1; lane 3, human α1(IV)NC1; and lane 4, rat α3(IV)NC1.
Characterization of chimeric human α3/α1(IV)NC1 domains
Chimeric human α3(IV)/α1(IV)NC1 domains were produced in order to locate the site of autoantibody binding within the α3(IV)NC1 domain. Substituting regions of human α1(IV)NC1 for corresponding regions of human α3(IV)NC1 provides a means of determining the critical sequences for antibody binding, since α1(IV)NC1 is not recognized by patients' sera. The NC1 domains of human α3(IV) and α1(IV) were divided into three regions, designated as amino, central and carboxyl regions, which were shuffled using the common restriction sites to manufacture six chimeric NC1 domains of α1(IV)/α3(IV). Each of these was subcloned into the expression vector pCMV 530 as described. The chimeras were defined according to the origins of their regions, e.g. α331 was derived from the amino and central regions of α3(IV)NC1 and the carboxyl region of α1(IV)NC1 (Fig. 1).
The binding of autoantibodies to the recombinant α3(IV)/α1(IV) chimeras was remarkably consistent (Fig. 4). All six sera showed the highest avidity for α331 and α333. Binding was not significant unless the chimera had the α3(IV) amino terminal region. The presence of an adjacent α3(IV)NC1 central region appeared to augment binding slightly. In contrast, the α3(IV) carboxyl region contributed very little to recognition; the reactivities of α333 and α331 were similar, whereas a striking contrast was observed in the reactivities of α333 and α133. A small degree of heterogeneity was observed in binding patterns between patients; two sera showed a moderate degree of reactivity for α113. Interestingly, the binding pattern observed with the MoAb P1 to the chimeric molecules was similar to that observed with patients' autoantibodies (not shown). Of the six chimeras, α331 was the only chimera bound by the MoAb W17 (Fig. 5). It was shown subsequently (see radiolabelling experiment) that constructs encoding α131 and α133 did not produce any detectable recombinant protein.
Fig. 4.
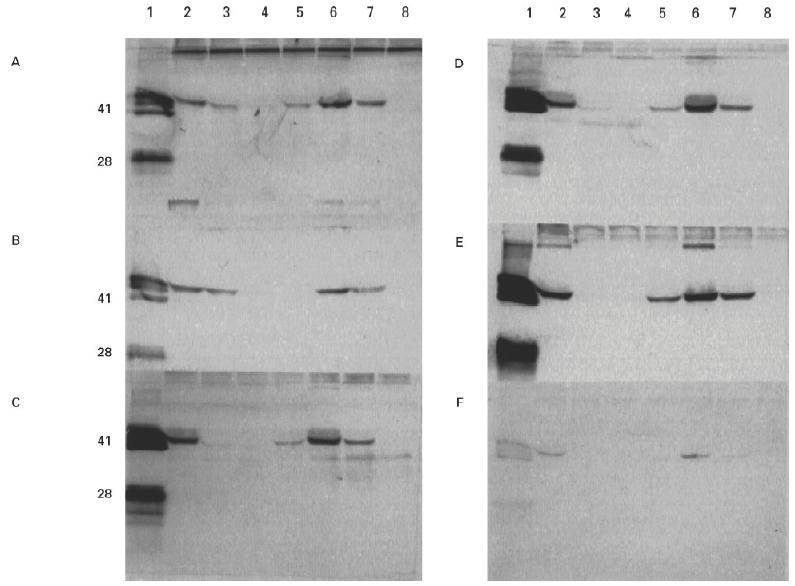
Western blots showing binding of human autoantibodies from six patients to chimeric α3/α1(IV)NC1 domains. COS supernatants from transfected cells were separated on 12.5% SDS–PAGE gels and electroblotted to nitrocellulose. Recombinant chimeric human α1(IV)/α3(IV)NC1 domains were detected with sera from Goodpasture patients A–F. Lane 1 contained collagenase-solubilized (cs) human glomerular basement membrane (GBM) as a positive control. The recombinant protein in the other lanes was: lane 2, human α3(IV)NC1 (α333); lane 3, α113; lane 4, α131; lane 5, α311; lane 6, α331; lane 7, α313; lane 8, α133.
Characterization of recombinant α331 chimera
As the α331 chimera appeared to have similar autoantibody binding properties to the recombinant Goodpasture antigen, α3(IV)NC1, it was decided to investigate its binding characteristics in greater detail. To this end, an equivalent volume of COS-7 cell supernatant containing recombinant α331 or α3(IV)NC1 was purified by cation exchange chromatography and concentrated on C30 columns (Amicon). By ELISA, the purified fraction of α331 showed a comparable degree of reactivity, with sera from patients with Goodpasture's disease, to that of recombinant α3(IV)NC1 and of native GBM (Fig. 6). Furthermore, α331 had the ability to inhibit the binding of Goodpasture autoantibodies to native human GBM, which was analogous to that of the recombinant antigen, α3(IV)NC1 (Fig. 7).
Fig. 6.
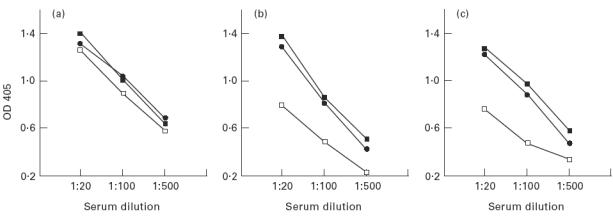
ELISAs comparing binding of sera from patients with Goodpasture's disease to human collagenase-solubilized (cs) glomerular basement membrane (GBM), recombinant α3(IV)NC1 and recombinant α331. Three patients' sera are shown in assays performed in parallel on ELISA plates coated with (a) 4 μg/ml of cs-human GBM, (b) 1:2 dilution of purified recombinant antigen, or (c) purified recombinant α331. Antigen preparations were diluted in carbonate buffer pH 7.5.
Fig. 7.
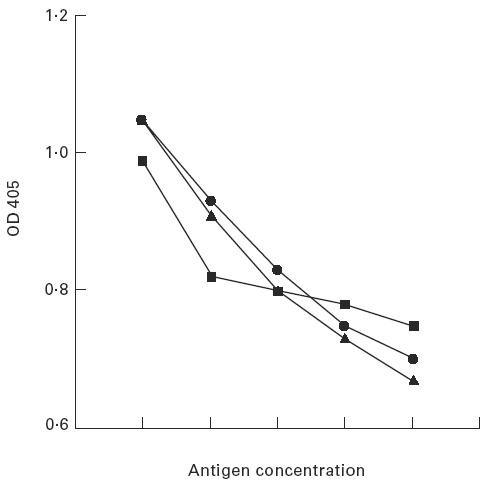
ELISA showing the inhibition of human autoantibodies binding to human glomerular basement membrane (GBM) by human GBM or recombinant α3(IV)NC1 or recombinant α331. Doubling amounts (arbitrary units) of human or purified human recombinant antigen or recombinant α331 were preincubated with patients' autoantibodies. These were then allowed to bind to collagenase-solubilized (cs) human GBM on an ELISA plate. ▪, Human cs-GBM; •, recombinant antigen; ▴, recombinant α331.
Characterization of chimeric human/rat α3(IV)NC1 domains
Since human autoantibodies recognize human α3(IV)NC1 considerably more strongly than rat α3(IV)NC1, it was reasoned that additional information may be obtained by studying chimeric forms of human/rat α3(IV)NC1. Rat α3(IV)NC1 has a 91% sequence homology with human α3(IV)NC1, compared with 75% for human α1(IV)NC1. Therefore, chimeras of human/rat α3(IV)NC1 domains were more likely to be conformationally similar to the intact recombinant human α3(IV)NC1. Thus, using an identical cloning strategy to that described above, the amino, central and carboxyl regions of human α3(IV)NC1 were sequentially substituted by the similar regions of rat α3(IV)NC1. The constructs were described according to the origins of their amino-terminal, middle and carboxyl-terminal regions, i.e. αHHR was derived from the amino-terminal and central regions of human α3(IV)NC1 and the carboxyl-terminal region of rat α3(IV)NC1. All recombinant chimeras bound human autoantibodies to some extent, but binding was strongest to chimeras containing the human α3(IV)NC1 amino region (Fig. 8). In contrast, all three chimeras with a rat α3(IV)NC1 amino region were bound weakly, irrespective of their central and carboxyl region composition. These results from the chimeric human/rat α3(IV)NC1 domain were entirely consistent with those from the chimeric human α3/α1(IV)NC1 domains. Both sets of data suggest that the amino terminal region of human α3(IV)NC1 is most important for the binding of autoantibodies. All the above experiments were repeated at least three times and consistently gave similar results.
Fig. 8.
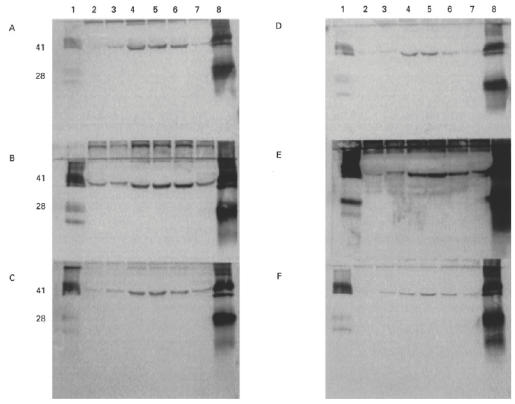
Western blots showing binding of human autoantibodies from six patients to chimeric human/rat α3(IV)NC1 domains. COS supernatants from transfected cells were separated on 12.5% SDS–PAGE gels and electroblotted to nitrocellulose. Recombinant chimeric human/rat α3(IV)NC1 domains were detected with sera from Goodpasture patients A–F. Lanes 1 and 8 contained, respectively, collagenase-solubilized (cs) rat glomerular basement membrane (GBM) and human GBM as positive controls. The recombinant protein in the other lanes was: lane 2, αRRH; lane 3, αRHR; lane 4, αHRR; lane 5, αHHR; lane 6, αHRH; lane 7, αRHH.
Quantification of chimeric α(IV)NC1 domains by 35S-radiolabelling
Since the observed differences in the reactivities of the chimeras with human autoantibodies could be due to quantitative differences in levels of expression, rather than qualitative differences in the immunoreactivity, the protein expression levels for all chimeras were compared. Seventy-two hours after transfection with the various chimeric constructs, COS-7 cell supernatants were replaced with cysteine- and methionine-free DMEM (−FCS) containing Promix (radiolabelled cysteine and methionine). The COS-7 cells were incubated for 6 h and supernatants were collected and run on SDS–PAGE gels. Immunoreactivities of chimeric proteins were assessed by Western blotting, and radioactive protein was detected by exposure of autoradiograph film to dried gels.
The assessment of expression levels for chimeric human α3(IV)/α1(IV) NC1 domains is shown in Fig. 9. The protein levels for four of the chimeras, and for recombinant α1(IV)NC1 and α3(IV)NC1, were comparable. However, there was no detectable expression of α131 and α133. The constructs for these chimeras were recloned and expressed, but a similar result was obtained. The assessment of expression levels for human/rat α3(IV)NC1 chimeras is shown in Fig. 10. For these, all six chimeras showed comparable levels of expression. The expression levels for rat α3(IV)NC1 and human α3(IV)NC1 were also comparable.
Fig. 9.
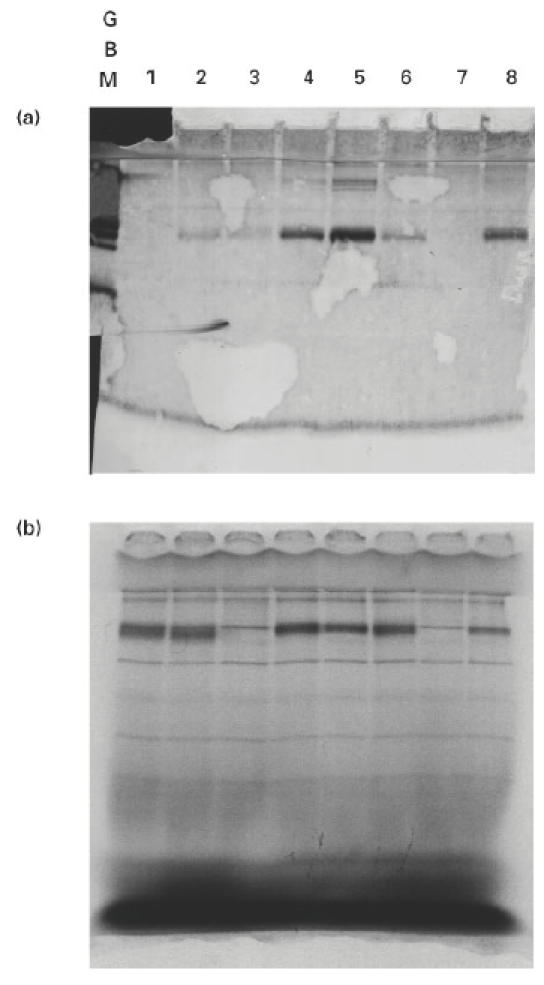
Western blot and autoradiograph of radiolabelled chimeric human α3(IV)/α1(IV)NC1 domains. COS supernatants from transfected cells were separated on 12.5% SDS–PAGE gels, which were then electroblotted to nitrocellulose or dried for exposure to autoradiograph film. (a) Western blot showing binding of Goodpasture autoantibodies to radiolabelled chimeric α3/α1(IV)NC1 domains. (b) Autoradiograph showing the expression levels of chimeric human α3(IV)/α1(IV)NC1 domains. The radiolabelled recombinant protein in each lane was: lane 1, human α1(IV)NC1 (α111); lane 2, α113; lane 3, α131; lane 4, α311; lane 5, α331; lane 6, α313; lane 7, α133; lane 8, human α3(IV)NC1 (α333).
Fig. 10.
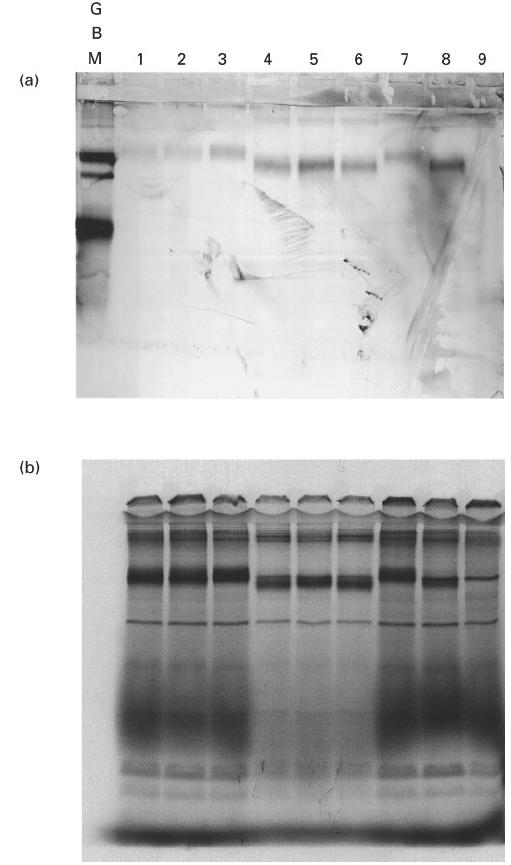
Western blot and autoradiograph of radiolabelled chimeric human/rat α3(IV)NC1 domains. COS supernatants from transfected cells were separated on 12.5% SDS–PAGE gels, which were then electroblotted to nitrocellulose or dried for exposure to autoradiograph film. (a) Western blot showing binding of Goodpasture autoantibodies to radiolabelled chimeric human/rat α3(IV)NC1 domains. (b) Autoradiograph showing the expression levels of chimeric human/rat α3(IV)NC1 domains. The radiolabelled recombinant protein in each lane was: lane 1, rat α3(IV)NC1 (α333); lane 2, αRRH; lane 3, αRHR; lane 4, αHRR; lane 5, αHHR; lane 6, αHRH; lane 7, αRHH; lane 8, human α3(IV)NC1; lane 9, negative control.
DISCUSSION
In this study we have produced strong evidence that the amino region of the Goodpasture antigen has a critical role in the binding of autoantibodies. We have used molecules related to human α3(IV)NC1 to create stable chimeric forms of the antigen. Recombinant human α1(IV)NC1, which has 74% amino acid sequence homology with human α3(IV)NC1, does not bind autoantibodies. Recombinant rat α3(IV)NC1, which has 91% homology, binds autoantibodies poorly. Our hypothesis was that substitution of regions of human α1(IV)NC1 and rat α3(IV)NC1 for corresponding regions in human α3(IV)NC1 would provide a means of determining the dominant regions of antigenicity within the Goodpasture antigen and thus map the major B cell epitope.
The COS cell transient expression system proved a very reliable and robust method for producing recombinant antigen for these studies. The system provided a means of rapidly expressing recombinant proteins at levels that were readily detectable in the COS supernatant. The results show that the construct previously used to produce conformationally correct antigen in insect cells [32] can be effective in other cell types. Most conformational epitopes are complex and are believed to contain five or six critical residues distributed across the molecule, or at least over a large region of it, which will only form the epitope ‘groove’ in the correctly folded protein [46]. Thus it is important to use experimental approaches which preserve the tertiary structure of the subject protein.
Other studies aimed at identifying the Goodpasture epitope have used two approaches. First, those using short synthetic peptides have produced conflicting results. One study claimed to have localized the major epitope by showing that synthetic peptides corresponding to sequences in the carboxyl region of the antigen were able to inhibit the binding of human autoantibodies to bovine α3(IV)NC1 monomer [28]. Our own studies, which examined the binding of Goodpasture antibodies to a panel of membrane-bound peptides spanning the entire antigen, failed to identify this region [29]. However, linear sequences identified by autoantibodies may contribute towards binding in a conformational epitope. It is also possible that antibodies binding to peptides in such studies were originally elicited by denatured protein, as a secondary response during the progress of the disease [46]. Second, a splicing variant of α3(IV)NC1, expressed in Escherichia coli, that retained the amino end of the molecule but lacked a large part of the carboxyl region, still bound strongly to human autoantibodies [30]. These observations are supported by a recent publication which suggested that the amino collagenous/NC1 region of α3(IV), together with the carboxyl region, played an important role in a major conformational epitope [47].
The production of recombinant chimeric molecules has proved to be a valuable tool in determining the regions of human α3(IV)NC1 critical for binding of human autoantibodies [48,49]. Results in the present study, using two sets of chimeras, were internally very consistent, in that the pattern of autoantibody binding to each set of six chimeras was similar for all six patients' sera examined. The MoAbs P1 and W17 demonstrated a binding pattern to the chimeras similar to that of patients' antibodies. It is interesting and important that both of these antibodies have previously been shown to be capable of blocking the binding of patients' autoantibodies to the Goodpasture antigen [35,45,50,51]. These observations demonstrate that the amino region of the α3(IV)NC1 domain is crucial for antibody recognition. One of the above mentioned studies also suggested that lysine residues in the carboxyl region were crucial for antibody recognition [28]. However, it would appear from our study that the absence of these ‘critical lysines’ from the carboxyl region of α1(IV)NC1 does not significantly alter the binding of autoantibodies to ‘conformationally correct’ recombinant α331 (Figs 6 and 7).
The assessment of the expression levels of the chimeric NC1 domains is interesting, and overall supports our interpretation of the Western blotting studies. It is difficult to say why there was no detectable expression of α131 and α133. It is interesting that the juxtaposition of the amino region of α1(IV)NC1 with the central region of α3(IV)NC1 is common to both chimeras. It could be speculated that this arrangement may result in some failure at the transcriptional or translational level, or in failure of secreting. Nevertheless, our results show that the central region of α3(IV)NC1 contributes very little to the immunoreactivity of the whole molecule; e.g. α313 and αHRH bind autoantibodies very well. It is also clear from these experiments that α113 has comparable expression levels to α333 or α331, but has a poor reactivity with autoantibodies. In addition, the counterparts of α131 and α133 in the experiment involving human/rat chimeras, namely αRHR and αRHH, have relatively weak reactivities with human autoantibodies. The overall interpretation of these studies is that the α3(IV)NC1 amino region is crucial for antibody reactivity, whereas the contribution of sequences in the carboxyl region is less important than had previously been thought. However, it is possible that amino acid residues from several parts of the molecule may contribute to antibody binding.
The information derived from this study of the properties of human α3(IV)/α1(IV)NC1 and human/rat α3(IV)NC1 chimeric molecules is consistent and has increased our understanding of the immunological properties of the autoantigen. The results should lay the foundations for further work on the relationship between B and T cell epitopes, and may be of value in the designing of new approaches to immunotherapy.
Acknowledgments
This work was supported by grants from the Medical Research Council, the National Kidney Research Fund and the Miranda Saunders Research Fund. N.T. was a NKRF Senior Research Fellow.
REFRENCES
- 1.Wilson CB, Dixon FJ. Anti-glomerular basement membrane antibody-induced glomerulonephritis. Kidney Int. 1973;3:74–89. doi: 10.1038/ki.1973.14. [DOI] [PubMed] [Google Scholar]
- 2.Glassock RJ. Natural history and treatment of primary proliferative glomerulonephritis: a review. Kidney Int Suppl. 1985;17:136–42. [PubMed] [Google Scholar]
- 3.Turner AN, Rees AJ. Goodpasture's disease and Alport's syndrome. Ann Rev Med. 1996;47:377–86. doi: 10.1146/annurev.med.47.1.377. [DOI] [PubMed] [Google Scholar]
- 4.Lerner RA, Glassock RJ, Dixon FJ. The role of anti-glomerular basement membrane antibody in the pathogenesis of human glomerulonephritis. J Exp Med. 1967;126:989–1004. doi: 10.1084/jem.126.6.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saus J, Wieslander J, Langeveld JPM, Quinones S, Hudson BG. Identification of the Goodpasture antigen as the α3 (IV) chain of collagen IV. J Biol Chem. 1988;263:13374–80. [PubMed] [Google Scholar]
- 6.Butkowski RJ, Langeveld JPM, Wieslander J, Hamilton J, Hudson BG. Localization of the Goodpasture epitope to a novel chain of basement membrane collagen. J Biol Chem. 1987;262:7874–7. [PubMed] [Google Scholar]
- 7.Gunwar S, Ballester F, Kalluri R, et al. Glomerular basement membrane. Identification of dimeric subunits of the noncollagenous domain (hexamer) of collagen IV and the Goodpasture antigen. J Biol Chem. 1991;266:15318–24. [PubMed] [Google Scholar]
- 8.Derry CJ, Pickering M, Baker C, Pusey CD. Identification of the Goodpasture antigen, α3 (IV) NC1, and four other NC1 domains of type IV collagen, by amino terminal sequence analysis of human GBM separated by 2-D electrophoresis. Exp Nephrol. 1994;2:249–56. [PubMed] [Google Scholar]
- 9.Hudson BG, Kalluri R, Gunwar S, Noelken ME, Mariyama M, Reeders ST. Molecular characteristics of the Goodpasture autoantigen. Kidney Int. 1993;43:135–9. doi: 10.1038/ki.1993.22. [DOI] [PubMed] [Google Scholar]
- 10.Siebold B, Deutzmann R, Kuhn K. The arrangement of intra- and intermolecular disulfide bonds in the carboxyterminal, non-collagenous aggregation and cross-linking domain of basement-membrane type IV collagen. Eur J Biochem. 1988;176:617–24. doi: 10.1111/j.1432-1033.1988.tb14321.x. [DOI] [PubMed] [Google Scholar]
- 11.Wieslander J, Bygren P, Heinegard D. Antiglomerular basement membrane antibody: antibody specificity in different forms of glomerulonephritis. Kidney Int. 1983;23:855–61. doi: 10.1038/ki.1983.106. [DOI] [PubMed] [Google Scholar]
- 12.Fish AJ, Lockwood MC, Wong M, Price RG. Detection of Goodpasture antigen in fractions prepared from collagenase digests of human glomerular basement membrane. Clin Exp Immunol. 1984;55:58–66. [PMC free article] [PubMed] [Google Scholar]
- 13.Hostikka SL, Tryggvason K. The complete primary structure of the α2 chain of human type IV collagen and comparison with the α1 (IV) chain. J Biol Chem. 1988;263:19488–93. [PubMed] [Google Scholar]
- 14.Sugimoto M, Oohashi T, Yoshioka H, Matsuo N, Ninomiya Y. cDNA isolation and partial gene structure of the human α4 (IV) collagen chain. FEBS Letters. 1993;330:122–8. doi: 10.1016/0014-5793(93)80256-t. [DOI] [PubMed] [Google Scholar]
- 15.Oohashi T, Sugimoto M, Mattei MG, Ninomiya Y. Identification of a new collagen IV chain, α6 (IV), by cDNA isolation and assignment of the gene to chromosome Xq22, which is the same locus for COL4A5. J Biol Chem. 1994;269:7520–6. [PubMed] [Google Scholar]
- 16.Hudson BG, Reeders ST, Tryggvason K. Type IV collagen: structure, gene organization, and role in human diseases. J Biol Chem. 1993;268:26033–6. [PubMed] [Google Scholar]
- 17.Soininen R, Huotari M, Ganguly A, Prockop DJ, Tryggvason K. Structural organization of the gene for the α1 chain of human type IV collagen. J Biol Chem. 1989;264:13565–71. [PubMed] [Google Scholar]
- 18.Morrison KE, Mariyama M, Yang-Feng TL, Reeders ST. Sequence and localization of a partial cDNA encoding the human α3 chain of type IV collagen. Am J Hum Genet. 1991;49:545–54. [PMC free article] [PubMed] [Google Scholar]
- 19.Turner N, Mason PJ, Brown R, et al. Molecular cloning of the human Goodpasture antigen demonstrates it to be the α3 chain of type IV collagen. J Clin Invest. 1992;89:592–601. doi: 10.1172/JCI115625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reeders ST. Molecular genetics of heriditary nephritis. Kidney Int. 1992;42:783–92. doi: 10.1038/ki.1992.348. [DOI] [PubMed] [Google Scholar]
- 21.Cashman SJ, Pusey CD, Evans DJ. Extraglomerular distribution of immunoreactive Goodpasture antigen. J Pathol. 1988;155:61–70. doi: 10.1002/path.1711550110. [DOI] [PubMed] [Google Scholar]
- 22.Kleppel MM, Santi PA, Cameron JD, Wieslander J, Michael AF. Human tissue distribution of novel basement membrane collagen. Am J Pathol. 1989;134:813–25. [PMC free article] [PubMed] [Google Scholar]
- 23.Kalluri R, Wilson CB, Weber M, et al. Identification of the α3 chain of type IV collagen as the common antigen in antibasement membrane disease and Goodpasture syndrome. J Am Soc Nephrol. 1995;6:1178–85. doi: 10.1681/ASN.V641178. [DOI] [PubMed] [Google Scholar]
- 24.Dehan P, Weber M, Zhang X, Reeders ST, Foidart J-M, Tryggvason K. Sera from patients with anti-GBM nephritis including Goodpasture syndrome show heterogenous reactivity to recombinant NC1 domain of type IV collagen α chains. Nephrol Dialysis Transplant. 1996;11:2215–22. doi: 10.1093/oxfordjournals.ndt.a027139. [DOI] [PubMed] [Google Scholar]
- 25.Lin RH, Mamula MJ, Hardin JA, Janeway Jr CA. Induction of autoreactive B cells allows priming of autoreactive T cells. J Exp Med. 1991;173:1433–9. doi: 10.1084/jem.173.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanzavecchia A. Receptor-mediated antigen uptake and its effect on antigen presentation to class II-restricted T lymphocytes. Annu Rev Immunol. 1990;8:773–93. doi: 10.1146/annurev.iy.08.040190.004013. [DOI] [PubMed] [Google Scholar]
- 27.Watts C, Lanzavecchia A. Suppressive effect of antibody on processing of T cell epitopes. J Exp Med. 1993;178:1459–63. doi: 10.1084/jem.178.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalluri R, Gunwar S, Reeders ST, et al. Goodpasture syndrome. Localization of the epitope for the autoantibodies to the carboxyl-terminal region of the α3 (IV) chain of basement membrane collagen. J Biol Chem. 1991;266:24018–24. [PubMed] [Google Scholar]
- 29.Levy JB, Coulthart A, Pusey CD. Mapping B cell epitopes in Goodpasture's disease. J Am Soc Nephrol. 1997;8:1698–705. doi: 10.1681/ASN.V8111698. [DOI] [PubMed] [Google Scholar]
- 30.Penades JR, Bernal D, Revert F, et al. Characterisation and expression of multiple alternatively spliced transcripts of the Goodpasture antigen gene region. Eur J Biochem. 1996;229:754–60. doi: 10.1111/j.1432-1033.1995.tb20524.x. [DOI] [PubMed] [Google Scholar]
- 31.Ryan JJ, Katbamna I, Mason PJ, Pusey CD, Turner AN. Sequence of the ‘Goodpasture antigen’ of different mammals. Nephrol Dial Transplant. 1998;13:602–7. doi: 10.1093/ndt/13.3.602. [DOI] [PubMed] [Google Scholar]
- 32.Turner N, Forstova J, Rees A, Pusey CD, Mason PJ. Production and characterisation of recombinant Goodpasture antigen in insect cells. J Biol Chem. 1994;269:17141–5. [PubMed] [Google Scholar]
- 33.Katbamna I, Turner AN, Ross C, Pusey CD. Development and application of an ELISA for Goodpasture's disease based on sheep α3 (IV) NC1 domains. J Clin Lab Immunol. 1995;47:11–24. [PubMed] [Google Scholar]
- 34.Derry CJ, Pusey CD. Specificity of autoantibodies in Goodpasture's syndrome on 2-D Western blotting of glomerular and alveolar basement membrane. Nephrol Dial Transplant. 1990;5:305. (Abstr.) [Google Scholar]
- 35.Pusey CD, Dash A, Kershaw MJ, et al. A single autoantigen in Goodpasture's syndrome identified by a monoclonal antibody to human glomerular basement membrane. Lab Invest. 1987;56:23–31. [PubMed] [Google Scholar]
- 36.Johansson C, Butkowski R, Wieslander J. Characterisation of monoclonal antibodies to the globular domain of type IV collagen. Connect Tiss Res. 1991;25:229–41. doi: 10.3109/03008209109029159. [DOI] [PubMed] [Google Scholar]
- 37.Bowman C, Peters DK, Lockwood CM. Anti-glomerular basement membrane autoantibodies in the Brown Norway rat: detection by a solid-phase radioimmunoassay. J Immunol Methods. 1983;61:325–33. doi: 10.1016/0022-1759(83)90227-2. [DOI] [PubMed] [Google Scholar]
- 38.Sambrook J, Fritsch EF, Maniatis T. 2. Cold Spring Harbor: Cold Spring Harbor Laboratory, A laboratory manual; 1989. Molecular cloning. [Google Scholar]
- 39.Mason PJ, Vulliamy TJ, Foulkes NS, Town M, Haidar B, Luzzatto L. The production of normal and variant human glucose-6-phosphate dehydrogenase in cos cells. Eur J Biochem. 1988;178:109–13. doi: 10.1111/j.1432-1033.1988.tb14435.x. [DOI] [PubMed] [Google Scholar]
- 40.Pihlajaniemi T, Tryggvason K, Myers JC, et al. cDNA clones coding for the pro-α1 (IV) chain of human type IV procollagen reveal an unusual homology of amino acid sequences in two halves of the carboxyl-terminal domain. J Biol Chem. 1985;260:7681–7. [PubMed] [Google Scholar]
- 41.Ryan JJ, Mason PJ, Turner AN, Pusey CD. Cloning, sequencing and expression of the NC1 domain of rat α3 (IV) collagen. J Am Soc Nephrol. 1996;7:1718. (Abstr.) [Google Scholar]
- 42.Chisholm V. High efficiency gene transfer into mammalian cells. In: Glover DM, Hames BD, editors. DNA cloning 4: Mammalian systems. 2. Oxford: IRL Press; 1995. pp. 16–19. [Google Scholar]
- 43.Blake MS, Johnston KH, Russell-Jones GJ, Gotschlich RC. A rapid, sensitive method for detection of alkaline phosphatase-conjugated anti-antibody on Western blots. Anal Biochem. 1984;136:175–9. doi: 10.1016/0003-2697(84)90320-8. [DOI] [PubMed] [Google Scholar]
- 44.Wheeler J, Sussman M. Enzyme-linked immunosorbent assay for circulating anti-glomerular basement membrane antibodies. Clin Exp Immunol. 1981;45:271–8. [PMC free article] [PubMed] [Google Scholar]
- 45.Derry CJ, Dunn MJ, Rees AJ, Pusey CD. Restricted specificity of the autoantibody response in Goodpasture's syndrome demonstrated by two-dimensional Western blotting. Clin Exp Immunol. 1991;86:457–63. doi: 10.1111/j.1365-2249.1991.tb02953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laver WG, Air GM, Webster RG, Smith-Gill SJ. Epitopes on protein antigens: misconceptions and realities. Cell. 1990;61:553–6. doi: 10.1016/0092-8674(90)90464-p. [DOI] [PubMed] [Google Scholar]
- 47.Kalluri R, Sue MJ, Hudson BG, Neilson EG. The Goodpasture antigen: structural delineation of two immunologically privileged epitopes on α3 (IV) chain of type IV collagen. J Biol Chem. 1996;271:9062–8. doi: 10.1074/jbc.271.15.9062. [DOI] [PubMed] [Google Scholar]
- 48.Ryan JJ, Mason PJ, Pusey CD, Turner AN. Chimaeric forms of type IV collagen NC1 domains used to analyse the Goodpasture antigen. Immunology. 1995;86 (Abstr.) [Google Scholar]
- 49.Ryan JJ, Mason PJ, Pusey CD, Turner AN. Identification of B cell epitopes of the Goodpasture antigen using chimaeric type IV collagen NC1 domains. Nephrol Dial Transplant. 1996;11:16. (Abstr.) [Google Scholar]
- 50.Hellmark T, Johansson C, Wieslander J. Characterisation of anti-GBM antibodies involved in Goodpasture's syndrome. Kidney Int. 1995;46:823–9. doi: 10.1038/ki.1994.338. [DOI] [PubMed] [Google Scholar]
- 51.Levy JB, Turner AN, George AJT, Pusey CD. Epitope analysis of the Goodpasture antigen using a resonant mirror biosensor. Clin Exp Immunol. 1997;106:79–85. doi: 10.1046/j.1365-2249.1996.d01-815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]