

Abstract
Human lung cancer expresses cell membrane complement inhibitory proteins (CIP). We investigated whether human lung cancer cell lines also express cell-membrane CIP molecules and whether the biology of CIP molecules in these cell lines differs from that of CIP in normal human respiratory epithelium in culture. The cell lines ChaGo K-1 and NCI-H596 were compared with normal human nasal epithelium in primary cultures in respect to the level of cell membrane CIP expression of membrane cofactor protein (MCP; CD46), decay-accelerating factor (DAF; CD55) and CD59, in respect to the level of cell resistance to complement-mediated lysis, and in respect to the contribution of cell membrane CIP to cell resistance against complement-mediated lysis. We found, using flow cytometry, that both human lung cancer cell lines expressed MCP, DAF and CD59, as did normal nasal epithelial cells. However, normal cells showed a large subpopulation of low DAF-expressing cells (60% of all cells) and a smaller subpopulation of high DAF-expressing cells (40%), while the lung cancer cell lines showed only one cell population, of high DAF expression. In addition, both lung cancer cell lines expressed higher MCP levels, and NCI-H596 cells showed higher levels of CD59. Cell resistance to complement-mediated lysis of both lung cancer cell lines was much higher than that of normal cells. Fifty percent normal human serum, under the same concentrations of complement activators, induced lysis of less than a mean of 10% of lung cancer cells, while lysing up to a mean of 50% of nasal epithelial cells. Lung cancer cell resistance to complement was due to its ability to prevent significant activation of complement upon its cell membrane, as manifested by a failure of complement activators to increase cell membrane deposition of C3-related fragments. The exact mechanism for this resistance remains obscure. Unexpectedly, neutralizing antibodies, anti-MCP and anti-DAF were entirely ineffective and anti-CD59 was only slightly effective (18% mean cell lysis) in increasing the susceptibility of the lung cancer cell lines to complement, while the same antibodies were very effective in facilitating complement-mediated lysis of the normal nasal epithelial cells (50% mean cell lysis with CD59 MoAb). On the other hand, detachment of DAF and CD59 by phosphatidylinositol-specific phospholipase C (PIPLC) from the lung cancer cell lines abrogated their resistance to lysis. We suggest that the biology of cell membrane CIP molecules in human lung cancer cell lines is different from that of CIP in normal respiratory epithelial cells. Human lung cancer cell lines are able to prevent significant complement activation upon its cell membrane and are therefore especially resistant to complement-mediated lysis. Complement resistance may serve this common and highly lethal human cancer as an escape mechanism from the body's immunosurveillance and prevent effective immunotherapy with tumour-specific MoAbs.
Keywords: CD46, CD55, CD59, complement inactivators, nasal epithelium, lung cancer cell lines, phosphatidylinositol-specific phospholipase C
INTRODUCTION
Normal human cells are usually protected from being lysed by homologous serum complement due to cell-membrane complement inhibitory proteins (CIP) [1,2].
These cell membrane CIP molecules act at different levels of the complement cascade and limit the activation of complement upon the cell membrane, which otherwise could result in formation of complement terminal membrane attack complexes that mediate cell lysis.
Recently we reported several new observations regarding the human respiratory tract and cell membrane regulation of complement. We reported that cell membrane CIP are expressed in respiratory epithelial cells along the entire human respiratory tract, in health and in disease, and that expression increases during inflammation and in lung cancer [3]. These cell membrane CIP are membrane cofactor protein (MCP; CD46), decay-accelerating factor (DAF; CD55) and CD59. In addition, we reported that complement activation may occur upon nasal respiratory epithelial cells in vivo [4], and that cell membrane CIP are essential for the protection of human nasal epithelial cells from complement-mediated lysis in vitro [5]. However, we showed that when complement activation surpasses the protective capacity of cell membrane CIP, normal nasal epithelial cells are lysed to a significant extent.
If cancer cells could selectively be lysed by homologous complement [6], cancer growth and metastases would be hampered. Unfortunately, many types of cancer cells were shown to express, similarly to normal cells, these cell membrane CIP and may therefore escape complement-mediated lysis [7,8], or even lysis by natural killer (NK) cells [9].
In the present study we investigate whether the biology of cell membrane CIP molecules in human lung cancer cell lines differs from that of CIP in normal human respiratory epithelium. We inquire first, whether cell membrane CIP are expressed in human lung cancer cell lines, and if so, whether it differs from expression of CIP in normal respiratory epithelium in culture. Second, whether cell resistance to complement-mediated lysis is different in lung cancer cell lines with respect to normal cells, and if so, what is the mechanism(s) that is responsible for this difference in resistance.
To answer these questions we studied both human lung cancer cell lines (bronchogenic carcinoma) and normal, non-cancer, human nasal respiratory epithelial cells in culture. We first assessed expression of cell membrane CIP by flow cytometry. Then we evaluated cell resistance to complement-mediated lysis in the presence and absence of complement activators, and also the effect of neutralizing antibodies against MCP, DAF and CD59 on cell resistance to complement-mediated lysis. Since we found that human lung cancer cell lines exhibit enchanced resistance to complement-mediated lysis, we tried to clarify some of the mechanisms responsible for this enhanced resistance. To do this we investigated, first, whether serum complement can be activated upon the cell membrane of these lung cancer cells, and second, whether MCP, DAF and CD59 contribute to inhibition of complement activation upon the lung cancer cell membrane. In some of the comparisons we made we used data that we have recently obtained for normal nasal epithelial cells in culture in a parallel study done concomitantly with this one [5].
MATERIALS AND METHODS
Cell culture reagents
Human placental collagen type IV (Sigma Type VI), bovine serum albumin (BSA; fraction V), DL-dithiothreitol, Dulbecco's PBS, crude collagenase from Clostridium histolyticum (type I) and magnesium-free Krebs–Henseleit solution, were all purchased from Sigma Chemical Co. (St Louis, MO). DME (4.5 g glucose/l), Ham's F12, l-glutamine, Eagle's minimum essential medium (MEM) containing non-essential amino acids, RPMI medium, fetal calf serum (FCS) and trypsin-EDTA solution were all purchased from Biological Industries (Beit-Haemek, Israel). Fetal bovine serum (FBS) was purchased from Gibco BRL, Life Technologies (Paisley, UK).
Human lung cancer cell lines
Human lung cancer non-small cell (bronchogenic carcinoma) cell lines were purchased from American Type Culture Collection (ATCC; Rockville, MD). We studied cell lines ChaGo K-1 (ATCC no. HTB 168, human undifferentiated bronchogenic carcinoma) and NCI-H596 (ATCC no. HTB 178, human adenosquamous bronchogenic carcinoma). Cells were grown in uncoated 25-cm2 culture flasks (Nunc, Roskilde, Denmark), seeded at 40 × 103/cm2 for ChaGo K-1 and 20 × 103/cm2 for NCI-H596.
The culture medium was RPMI 1640 supplemented with 10% FBS and antibiotics (penicillin 100 U/ml, and streptomycin 100 μg/ml). Medium was renewed every 3 days. Cell cultures reached confluency after 5–7 days.
Human nasal epithelium primary cell cultures
Human normal nasal epithelial cells were isolated from freshly resected inferior nasal turbines. Nasal turbines were resected according to strict medical indications after a written informed consent was given by the patient. Patients suffered from atopic rhinitis or vasomotor rhinitis with persistent nasal obstruction. Cell isolation and cell culturing were performed similarly to the way we previously described [3]. Briefly, nasal turbines were first washed extensively and then incubated with a collagenase digesting solution for two periods, one of 60 min and a second of 120 min. Isolated cells were then seeded at a concentration of 4.5 × 105 cells/cm2 in 25-cm2 culture flasks that were precoated with human placental collagen type IV (25 μg/cm2). Cell cultures were incubated at 37°C in a water-saturated atmosphere of 5% CO2, in air. Feeding medium was changed after 24 h and then every 48 h. It was composed of DME mixed with Ham's F12 in a ratio of 1:1 and contained the following supplements: FCS (5%, v/v), non-essential amino acids in Eagle's MEM (1%), penicillin 100 U/ml, streptomycin 100 μg/ml, fungizone 0.5 μg/ml, and gentamycin 50 μg/ml. Cell cultures became confluent after 3–4 days, had a cobblestone appearance, and were used in experiments on days 4 and 5. The epithelial nature of cells cultured in this way has been verified by intense staining with anti-keratin MoAb (not shown).
Antibodies and human sera
Mouse anti-human MCP MoAb GB-24 (IgG 1,K) [10] was purchased from Theramex (Monaco). Mouse anti-human DAF MoAb IA10 (IgG2a) [11] was a kind gift of Dr V. Nussenzweig and Dr M. B. Whitlow (NYU Medical Center, New York, NY). Rat anti-human CD59 MoAb YTH 53.1 (IgG2b) was purchased from Serotec (Oxford, UK) [12]. Polyclonal antiserum to human nasal epithelial cells (anti-NEC) was prepared as previously described [5] by immunizing a rabbit with these cells. Smears of cultured nasal epithelial cells and lung cancer cells were positively stained with a 1:50 dilution of this antiserum using the streptavidin-biotin immunoperoxidase method. Anti-human carcinoembryonic antigen (anti-CEA, purified immunoglobulin fraction of rabbit antiserum) was purchased from Dako (Glostrup, Denmark). Anti-human C3c, FITC-conjugated (purified immunoglobulin fraction of rabbit antiserum) for flow cytometry studies was purchased from Dako. It should be noted that anti-human C3c serum identifies C3 native protein as well as its related fragments, produced during its activation, C3b, iC3b and C3c. FITC-conjugated goat serum anti-human IgD was purchased from Kallestad Labs (Chaska, MN). FITC-conjugated secondary F(ab′)2 antibodies for flow cytometry studies were purchased from Jackson Immunoresearch Labs (West Grove, PA). Normal human serum (NHS) was prepared from blood of normal non-smoking volunteers. Aliquots of NHS were kept at −70°C until use.
Expression of cell membrane CIP
In order to evaluate expression of cell membrane CIP in cancer and non-cancer cells of human respiratory tract epithelium, we assayed expression of MCP, DAF and CD59 in ChaGo K-1 and NCI-H596 human bronchogenic carcinoma cell lines and in non-cancer human nasal epithelial cells in primary cultures, by flow cytometry analysis. Confluent cultures of cancer and non-cancer cells were first rinsed with 2 ml of 0.25% trypsin–EDTA solution and then placed in a cell culture incubator for 5–7 min. Cells were then aspirated and mechanically dispersed with a glass pipette and extensively washed by PBS containing 10% heat-inactivated FCS. Cell viability by trypan blue was always > 96% for all cell types treated by trypsin–EDTA.
Cells were resuspended in ‘enriched’ PBS (containing calcium chloride and magnesium sulfate 1 mm each, BSA and glucose (0.5% each)), and were incubated with primary antibodies for the various cell membrane CIP (30 min, 4°C). Cells were then washed twice in ‘enriched’ PBS, and incubated with secondary antibodies, goat F(ab′)2 anti-rat IgG (H + L) and goat F(ab′)2 anti-mouse IgG (H + L), both FITC-conjugated (30 min, 4°C). Cells were then extensively washed and resuspended in ice-cold PBS (0.5 ml) at a cell concentration of 106/ml and analysed for fluorescence by a flow cytometer (Epics Profile II Flow Cytometer; Coulter Electronics, Luton, UK). The flow cytometer was equipped with an argon ion laser, using 15 mW light with an excitation wave length of 488 nm and detection wave length of 525 nm. The voltage used was 850 V. Five thousand cells were analysed by gating on a uniform cell population on a two-parameter histogram of forward versus side scatter. The fluorescence histograms were overlaid to determine significant differences from negative control antibodies. For negative control antibodies we used the appropriate FITC-conjugated secondary antibodies, or FITC-conjugated goat serum anti-human IgD (Kallestad Labs).
In preliminary studies we first determined the optimal dilution of primary antibodies needed in order to saturate each type of cell membrane CIP on each cell type. We used these optimal dilutions for our further assays. In order to compare mean fluorescence intensities (MFI) for each type of CIP of nasal epithelial cells with those of lung cancer cells, we corrected the MFI for the different mean cell surface area of each cell type. Cell diameter of suspended cells was used as an indicator for cell surface area. To determine the mean diameter for each cell type, a drop of cell suspension was placed on a glass slide and photographed under the microscope. Photograph negatives were then projected by a slide projector on a wall from a fixed distance and the diameter of 100 cells of each cell type from two different cell cultures was measured in millimetres and mean cell diameter (MCD) was calculated. The MCD of non-cancer nasal epithelial cells was considered as a reference and the MCD of each lung cancer cell line was calculated as its ratio: MCD ratio for a specific cancer cell line = MCD cancer divided by MCD non-cancer. To obtain the corrected, relative MFI for each cancer cell type, the mean of its MFI was divided by its MCD ratio.
Cell lysis assay protocol
For all lysis assays, we used single-cell suspensions obtained from confluent cultures of epithelial cells. Single-cell suspensions were prepared by trypsin–EDTA solution as described for flow cytometry analysis.
The lysis assay was performed in two steps. In the first step 5 × 105 epithelial cells were treated with diluted antibodies or antisera to cell membrane CIP, to NEC or to CEA at optimal dilutions in 100 μl ‘enriched’ PBS for 30 min at 4°C with gentle rotatory shaking. Control cells were treated with PBS alone, with non-immune mouse IgG (diluted 1:50 in PBS) and with heat-inactivated normal rabbit serum (diluted 1:20 in PBS) when appropriate. The cells were then washed twice with ‘enriched’ PBS at room temperature.
In the second step of the lysis assay, the cells were resuspended in 100 μl of 50% NHS in ‘enriched’ PBS with or without Escherichia coli lipopolysaccharide (LPS; serotype 026:B6; Sigma) and were gently shaken in a water-bath for 60 min at 37°C. Control cells were treated with PBS alone. Cells were then washed with PBS and resuspended in 0.2% trypan blue in PBS. Cell lysis was determined microscopically by the trypan blue dye inclusion technique. In preliminary studies we compared this method with a method using propidium iodide and flow cytometry and found these methods to be highly intercorrelated. A total of 300 cells was counted for each test tube and the percentage of cells containing dye was calculated. The percentage of cells lysed was calculated by subtracting the percentage of dye-containing cells in a PBS control assay from the percentage of dye-containing cells in the experimental assay.
Cell resistance to complement-mediated lysis
To evaluate the resistance of human lung cancer cell lines to complement-mediated lysis we assayed cell lysis after exposure to various concentrations of NHS with or without the addition of potential complement activators at various concentrations. Complement activators that we used were LPS of E. coli and cell membrane immunocomplexes formed by antibodies against cell membrane antigens that are not related to complement regulation. These antibodies were anti-CEA and anti-NEC (obtained as specified in the antibodies subsection above). Flow cytometry studies revealed that 100% of both nasal epithelial cells and lung cancer cell lines reacted with anti-NEC and that 100% of both lung cancer cell lines reacted with anti-CEA. However, flow cytometry revealed two subpopulations of CEA-expressing cancer cells, one subpopulation with high CEA expression (30% of cells) and a second one with low CEA expression (70% of cells). This pattern was confirmed also by peroxidase immunocytochemistry (not shown). Results of lung cancer cell line resistance studies were compared with results of nasal epithelial cell resistance which we obtained recently in a parallel and concomitant study [5].
Effect of neutralizing antibodies against MCP, DAF and CD59 on cell susceptibility to complement-mediated lysis
To evaluate the effect of neutralizing antibodies against cell membrane CIP, lung cancer cell lines were incubated in the first step of the lysis assay with different dilutions of anti-MCP, anti-DAF and anti-CD59 MoAbs, each one alone and in various combinations. In the second step of the assay, cells were incubated with 50% NHS in PBS and cell lysis was then assessed. In some experiments, in the second step of the assay, LPS (10 μg/ml) or anti-CEA (1:20) were added.
The results of the effect of neutralizing antibodies against the cell membrane CIP of lung cancer cell lines were compared with the results of the effect of these same antibodies on cell membrane CIP of non-cancer nasal respiratory epithelium that we obtained in a recent parallel study [5]. In addition, since in our recent study CD59 was neutralized by an antiserum, in the present study we assayed the effect of MoAb YTH 53.1 anti-CD59 on nasal epithelial cells, as well as on lung cancer cells.
Complement activation upon the cell membrane of lung cancer cell lines
To investigate whether complement is activated upon the cell membrane of lung cancer cell lines, and to what extent, we assessed the cell membrane adsorption of complement C3-related fragments using flow cytometry after incubation of such cells with NHS, with or without the addition of complement activators. Complement activators were either LPS of E. coli, serotype 026:B6 (Sigma), or cell membrane immunocomplexes formed by presensitization of cells with anti-NEC serum and anti-CEA antibodies. Cells in suspension were incubated with PBS alone or with 1%, 3%, 5%, 10% and 50% NHS in PBS (37°C, 30 min). In some experiments LPS (10 μg/ml) was added to 50% NHS just before incubation with the cells. In other experiments cells were sensitized with anti-NEC serum or with anti-CEA antibodies (4°C, 30 min, 1:10–20 dilution), washed in PBS and then incubated with 50% NHS for 30 min. At the end of the incubation time cells were washed twice in PBS and incubated at 4°C for 30 min with FITC-conjugated rabbit antibodies to human C3c (1:20).
Contribution of MCP, DAF and CD59 to inhibition of complement activation and to cell resistance
Neutralizing antibodies
MCP and DAF are known to interfere in the early steps of complement activation involving complement C3 in both pathways of the complement cascade. To determine whether MCP and DAF are essential for the inhibition of complement activation upon the cell membrane of lung cancer cell lines, we assessed cell membrane adsorption of complement C3-related fragments after treating the cancer cells with anti-MCP and anti-DAF neutralizing antibodies. Cancer cells were first incubated with anti-MCP and anti-DAF (4°C, 30 min) at saturating dilutions (1:50 as determined by flow cytometry). Cells were then washed in PBS and incubated with NHS without or with the addition of LPS.
In other experiments, before incubation with NHS, cells were sensitized with anti-NEC serum and with anti-CEA antibodies. Cells were then washed with PBS and flow cytometry for C3-related fragments was carried out as described above.
Phosphatidylinositol-specific phospholipase C treatment
Another approach that was taken in order to determine whether DAF and CD59 contribute to the resistance of the lung cancer cell lines to complement-mediated lysis was to detach DAF and CD59 from the cell membrane of these cells and to then reassess the cells’ susceptibility to lysis. Detachment of DAF and CD59 was obtained by treating the cells with phosphatidylinositol-specific phospholipase C (PIPLC) which acts on glycosyl-phosphatidylinositol (GPI) cell membrane anchors. PIPLC was kindly provided by Dr M. G. Low (Columbia University New York, NY). PIPLC was isolated from culture supernatants of Bacillus subtilis transfected with the PIPLC gene from B. thuringiensis [13,14]. In preliminary experiments we determined by flow cytometry that 0.5 U/ml of PIPLC (37°C, 60 min) detaches almost 100% of DAF and CD59 from both cancer cell lines, while MCP is untouched. To determine the complement lysis threshold after complete detachment of DAF and CD59, the cancer cell lines were treated first with PIPLC (0.5 U/ml, 37°C, 60 min), then washed and incubated with various concentrations of NHS (37°C, 60 min). In order to determine the threshold of DAF and CD59 detachment required for obtaining complement-mediated cell lysis, the lung cancer cell lines were first treated with various concentrations of PIPLC (0.0001–0.5 U/ml), then washed and incubated with 50% NHS (37°C, 60 min).
After incubation with NHS, cells were washed twice and cell lysis was assessed microscopically by the trypan blue inclusion technique. A total of 300 cells was counted from each assay and the percentage of cells containing dye was calculated. Cell treatment with PIPLC in PBS (0.5 U/ml, 37°C, 45 min), PIPLC control assay, was done for each experiment and was not found to induce cell lysis in comparison with PBS alone (< 3% dead cells). Percentage of cell lysis was calculated by subtracting the percentage of dye-containing cells in a PIPLC control assay from that in a test assay.
Statistical analysis
Mean values of the various groups were compared by one-way analysis of variance (one-way anova) or by Student's t-test when appropriate and as indicated in the figure legends.
RESULTS
Expression of cell membrane CIP
Both human bronchogenic carcinoma cell lines, NCI-H596 and ChaGo K-1, expressed MCP, DAF and CD59, as did non-cancerous human nasal epithelial cells (Fig. 1a, b, c). However, using flow cytometry, two populations of non-cancer nasal epithelial cells expressing DAF were identified (Fig. 1d). One population (40% of all cells) showed a high MFI and the second (60% of cells) showed an MFI that was 2.8 times lower. In contrast, only one population of DAF-expressing cells was identified in either lung cancer cell line, and this was a cell population with a relatively high DAF expression (Fig. 1d).
Fig. 1.
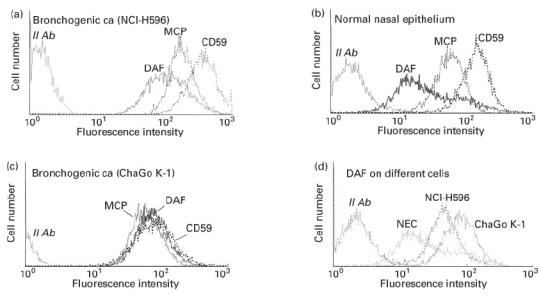
Cell membrane expression of complement inhibitory protein (CIP) molecules [membrane cofactor protein (MCP), decay-accelerating factor (DAF), CD59] as assessed by flow cytometry of human lung cancer cell lines (non-small cell bronchogenic carcinoma) and of normal nasal epithelial cells in primary cultures.
The relative expression of MCP, DAF and CD59 between cancer cell lines and non-cancer nasal epithelial cells was compared after correcting MFI for the MCD ratio for each cell type (see Materials and Methods). When the MCD of non-cancer nasal epithelial cells was considered as a reference of 1.0, MCD ratio of ChaGo K-1 and NCI-H596 cancer cell lines was 0.594 and 0.745, respectively. After such correction was done it was shown (Table 1) that both lung cancer cell lines expressed more cell membrane MCP and DAF than did non-cancer nasal respiratory epithelial cells. CD59 expression was higher in NCI-H596 and was lower in ChaGo K-1 cancer cell lines in respect of non-cancer nasal epithelial cells (Table 1).
Table 1.
*Corrected relative mean fluorescence intensity (MFI)† for human cell membrane complement inhibitory proteins (CIP) of lung cancer cells and of non-cancer nasal epithelial cells

Cell resistance to complement-mediated lysis
Both cell lines of human lung cancer were very resistant to complement-mediated lysis. Introducing complement activators like LPS (Fig. 2a, b) or cell membrane immunocomplexes, formed by presensitization of cells with anti-NEC or anti-CEA antibodies (Fig. 3), did not evoke a significant cell lysis. In contrast, non-cancer nasal epithelial cells were much less resistant and a high percentage of cells was lysed when LPS or anti-NEC were indroduced (Fig. 2c and Fig. 3).
Fig. 2.
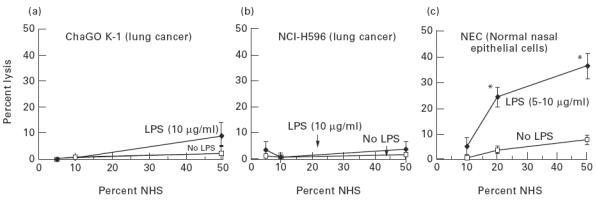
Effect of lipopolysaccharide (LPS) on threshold of susceptibility of human lung cancer cell lines (a,b) and normal nasal epithelial cells (c) to complement-mediated lysis. Cells were exposed to normal human serum (NHS) alone or in presence of LPS for 60 min at 37°C. Values are mean ± s.d. of six experiments. *P < 0.03 versus the preceding value (one-way anova). Data for normal nasal epithelial cells were obtained from our recent concomitant study [5].
Fig. 3.
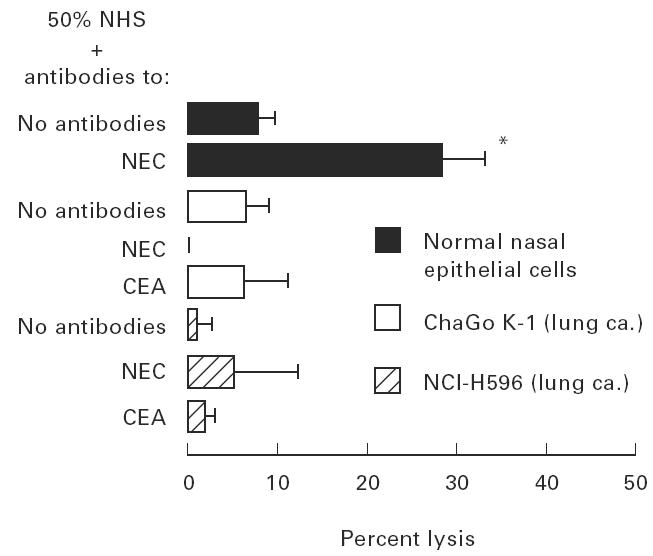
Effect of cell membrane immunocomplexes on complement-mediated lysis. Cells were presensitized with nasal epithelial cell (NEC) antiserum or with anti-carcinoembryonic antigen (CEA) (30 min, at 4°C). Cells were then washed and exposed to 50% normal human serum (NHS) (60 min, at 37°C). Values are mean ± s.d. of six experiments. *P < 0.0001 versus NEC with NHS and no antibodies (Student's t-test). Data for normal NEC were obtained from our recent concomitant study [5].
Effect of neutralizing antibodies against MCP, DAF and CD59 on cell susceptibility to complement-mediated lysis
Neutralizing antibodies against MCP, DAF and CD59 of both lines of lung cancer cells were not very effective in increasing susceptibility to complement-mediated lysis (Fig. 4a).
Fig. 4.
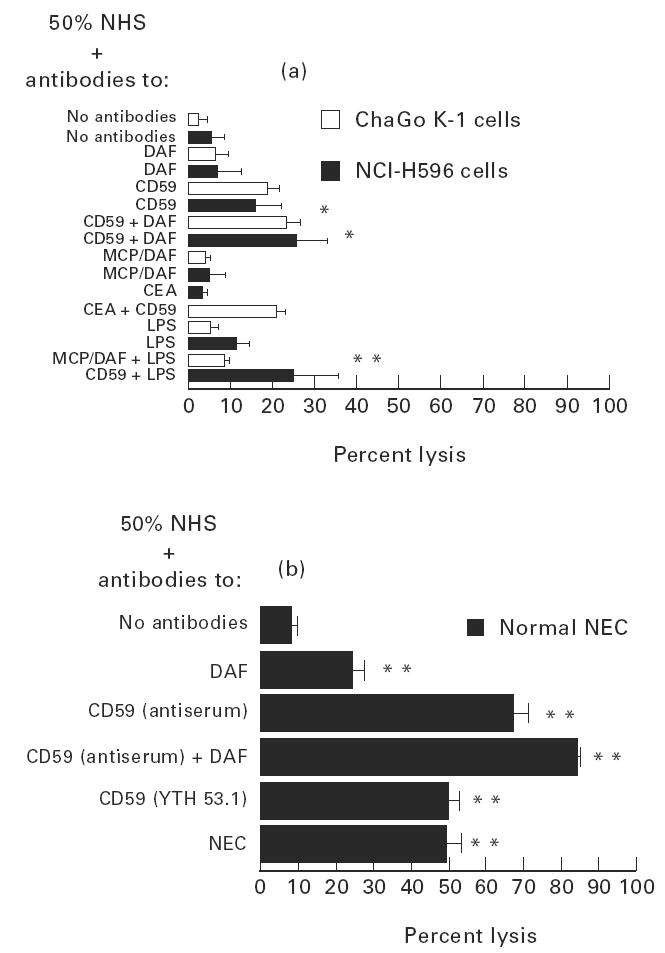
Effect of neutralizing antibodies against cell membrane complement inhibitory protein (CIP) molecules [anti-membrane cofactor protein (MCP), GB-24, decay-accelerating factor (DAF), IA-10, anti-CD59, YTH 53.1 or antiserum as indicated] on complement-mediated cell lysis of lung cancer cell lines (a) and of normal nasal epithelial cells (NEC) (b). Cells were incubated with antibodies (1:50 dilution, 30 min, at 4°C), then washed and exposed to 50% normal human serum (NHS). In some experiments cells were presensitized with anti-carcinoembryonic antigen (CEA) or with anti-NEC (1:20 dilution, each). Values are mean ± s.d. of four to six experiments. (a) *P < 0.01 versus lysis of same cells with NHS and no antibodies or versus lysis with anti-CD59; **P < 0.025 versus lysis of same cells with NHS and no antibodies. (b) **P < 0.001 versus lysis of same cells with NHS and no antibodies (Student's t-test for all comparisons). Data for normal NEC, except for the effect of anti-CD59, YTH 53.1, were obtained from our recent concomitant study [5].
Anti-MCP and anti-DAF were not effective at all. Anti-CD59 (YTH 53.1) was the only neutralizing antibody that increased complement-mediated lysis. Adding complement activators after treatment with neutralizing antibodies did not increase cell lysis further (Fig. 4a).
In contrast, anti-DAF and anti-CD59 were much more effective in increasing non-cancer nasal epithelial cell susceptibility to complement-mediated lysis (Fig. 4b). The effect of CD59 antiserum was especially pronounced, enabling lysis of up to a mean of 68% of nasal cells, but MoAb to CD59 (YTH 53.1) was also very effective, enabling lysis of up to a mean of 50% of these normal cells (Fig. 4b). In all cell types, cancer and non-cancer, the combined effect of DAF and CD59 was only additive (Fig. 4).
Mechanism of lung cancer cell lines’ resistance to complement
Complement activation upon the cell membrane of lung cancer cell lines
Complement C3-related fragments were adsorbed onto the cell membrane of both human lung cancer cell lines. The percentage of cells adsorbing C3-related fragments in the presence of NHS without the addition of complement activators increased proportionally as the concentration increased, reaching 100% of positive cells in the presence of 50% NHS, as shown by flow cytometry (Fig. 5).
Fig. 5.
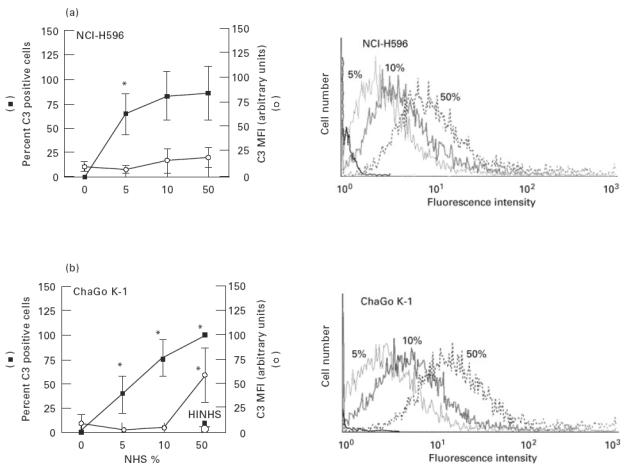
Spontaneous complement activation upon human non-small cell lung cancer cell lines (NCI-H596 and ChaGo K-1). Lung cancer cells were incubated with different concentrations of normal human serum (NHS) (37°C, 30 min), then washed in PBS and incubated with FITC-conjugated rabbit antibodies to human complement C3c (4°C, 30 min). Cells were then washed and analysed for cell membrane C3-related fragments by a flow cytometer. In some experiments ChaGo K-1 cells were incubated with 50% NHS that was first heat-inactivated (HINHS). (a) Percent of C3-positive cells and mean fluorescence intensity (MFI) are depicted. Values are mean ± s.d. of four (NCI-H596) and six to seven experiments (ChaGo K-1). *P < 0.01 versus the preceding value (one-way anova). (b) Examples of flow cytometry profiles for C3 deposition on the corresponding cell lines.
However, the amount of adsorbed C3-related fragments was low and did not increase with the increase of NHS concentration, except in ChaGo K-1 cells and only when 50% NHS was used (Fig. 5). Heat inactivation of 50% NHS completely abolished this C3 adsorption (Fig. 5). Introducing complement activators by adding LPS or by presensitization of ChaGo K-1 cells with anti-NEC or anti-CEA antibodies, even in the presence of anti-MCP and anti-DAF, before cell incubation with 50% NHS, did not affect cell membrane adsorption of C3-related fragments in a consistent and significant way (Fig. 6).
Fig. 6.
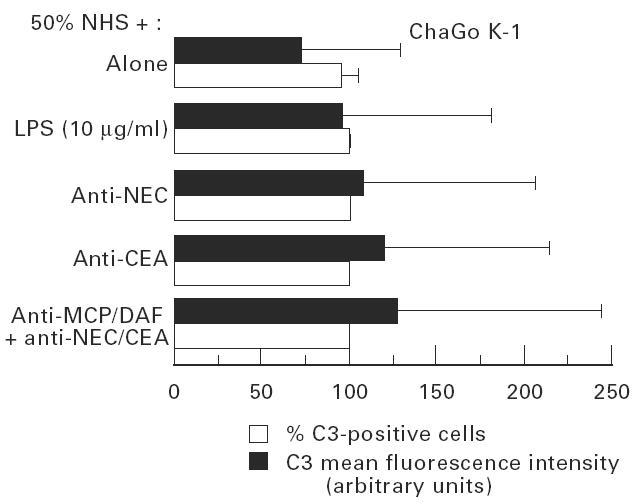
Complement activation upon ChaGo K-1 lung cancer cells in presence of complement activators in presence or absence of anti-membrane cofactor protein (MCP) or anti-decay-accelerating factor (DAF). Cells were incubated with 50% normal human serum (NHS). In some experiments complement activators were introduced. Lipopolysaccharide (LPS) (10 μg/ml) or cell membrane immunocomplexes were formed upon cancer cells by presensitization with anti-nasal epithelial cells (NEC) or anti-carcinoembryonic antigen (CEA) antibodies (1:10–20, 30 min, 4°C). In other experiments cells were first incubated with anti-MCP and/or anti-DAF (1:50, 30 min, 4°C) before presensitization with anti-NEC or anti-CEA. Cell membrane adsorption of C3-related frgments was assessed by flow cytometry using FITC-conjugated anti-human C3c. Values are mean ± s.d. of five experiments. Values were not significantly different from values of cells treated by NHS alone (Student's t-test).
Contribution of MCP, DAF and CD59 to inhibition of complement activation and to cell resistance
Neutralizing antibodies
Treatment of both lung cancer cell lines with neutralizing antibodies to MCP and DAF did not increase adsorption of C3-related fragments in the presence of 50% NHS (Fig. 7). When neutralizing antibodies to MCP and DAF were used in the presence of complement activators, cell membrane adsorption of C3-related fragments increased only to a minor degree (Fig. 6).
Fig. 7.
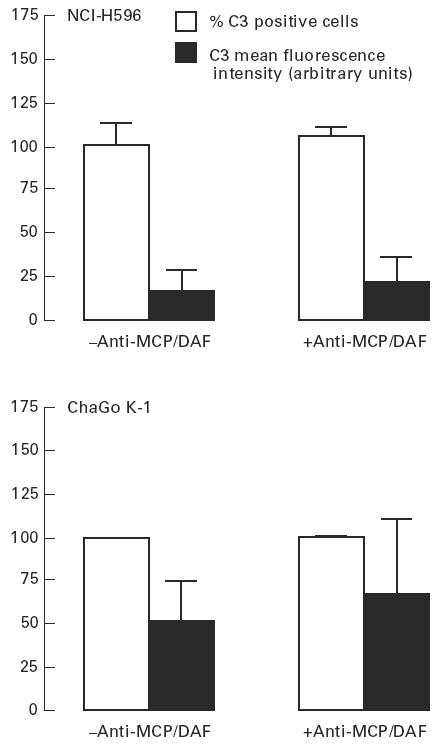
Effect of neutralizing antibodies against membrane cofactor protein (MCP) and decay-accelerating factor (DAF) on spontaneous complement activation upon lung cancer cell lines. Cells were incubated with 50% normal human serum (NHS) (37°C, 30 min). In some experiments cells were preincubated with anti-MCP and/or anti-DAF (1:50, 4°C, 30 min). Adsorption of C3-related fragments was assayed by flow cytometry. Values are mean ± s.d. of three (NCI-H596) and five (ChaGo K-1) experiments. Values of cells treated by anti-MCP/DAF were not significantly different from values of untreated cells (Student's t-test).
PIPLC treatment
PIPLC treatment, given to both lung cancer cell lines, detached DAF and CD59 completely while leaving MCP in place (Fig. 8), and increased cancer cell lysis in the presence of 50% NHS in a concentration-dependent manner (Fig. 9a). PIPLC at 0.5 U/ml (45 min, 37°C) facilitated complement-mediated lysis of > 80% of cells from both lines. When a fixed concentration of 0.5 U/ml PIPLC was used, NHS at 5% and 10% concentrations was insufficient to mediate a significant cell lysis and 50% NHS was required in order to mediate an extensive cell lysis (Fig. 9b).
Fig. 8.
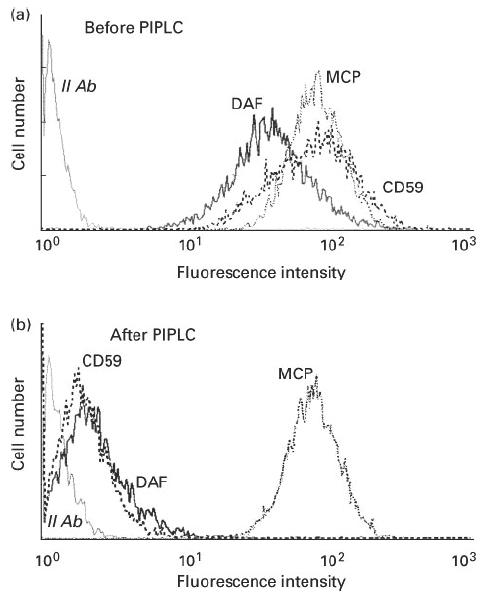
Detachment of decay-accelerating factor (DAF) and CD59 from the NCI-H596 lung cancer cell line cell membrane by Bacillus thuringiensis phosphatidylinositol-specific phospholipase C (PIPLC; 0.5 U/ml, 37°C, 45 min). Expression of cell membrane DAF and CD59 was assayed by flow cytometry. Results for ChaGo K-1 cells were the same (not shown).
Fig. 9.
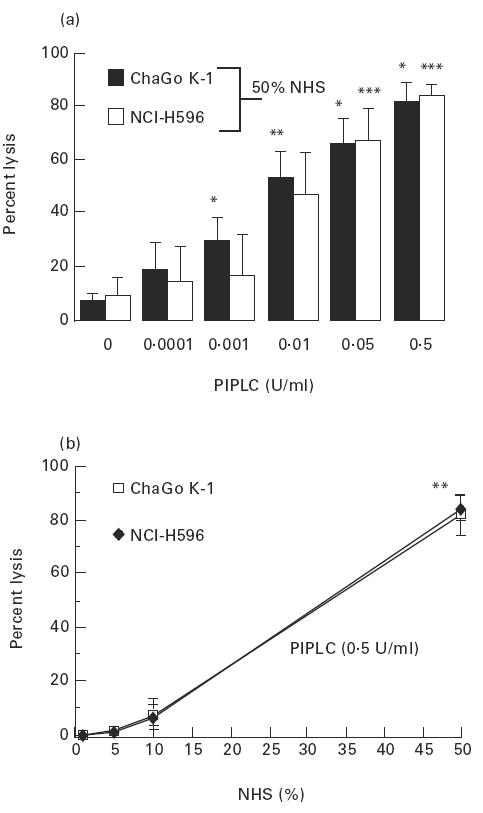
Effect of decay-accelerating factor (DAF) and CD59 detachment from the cell membrane of lung cancer cell lines on the cells’ susceptibility to complement-mediated lysis. (a) Cells were treated first with phosphatidylinositol-specific phospholipase C (PIPLC; 45 min, 37°C) at various concentrations, then washed and exposed to 50% normal human serum (NHS; 60 min, 37°C). *P < 0.001 from the alternate preceding value; **P < 0.05 from preceding value for ChaGo K-1 cells; ***P < 0.01 from the alternate preceding value, for NCI-H596 cells. (b) Cells were first treated with PIPLC, then washed and exposed to various concentrations of NHS. **P < 0.001 from the preceding value for both cell types. Values are mean ± s.d. of four to six experiments (one-way anova for all comparisons).
DISCUSSION
This study shows in a specific cell culture system that the biology of cell membrane CIP molecules of human lung cancer cell lines differs from that of normal, non-cancer human respiratory epithelium in culture.
CIP molecules, MCP, DAF and CD59 are expressed in lung cancer cell lines, but the level of expression is essentially higher than that of normal nasal epithelial cells. In addition, the lung cancer cell lines are much more resistant to complement-mediated lysis than are normal cells. We found that this increase in resistance is due to the ability of lung cancer cells to limit complement activation upon their cell membrane. As to the mechanism of this resistance, GPI-anchored cell membrane CIP molecules seem to be essential in preventing lung cancer cell line lysis by complement. Unexpectedly, however, we found that neutralizing antibodies against these cell membrane CIP have only a slight effect on the susceptibility of the lung cancer cell lines to complement-mediated lysis, while they significantly increase susceptibility to complement in non-cancer nasal epithelial cells.
The significance of cell membrane CIP molecules in the biology of human lung cancer was previously not investigated. Our finding that these molecules are expressed not only in vivo [3] but also in cell lines of human lung cancer, as was also shown by others [16], provides an in vitro model for studying the biological significance of human lung cancer CIP.
Expression of cell membrane MCP, DAF and CD59 in the lung cancer cell lines that we studied was, with the exception of CD59 in ChaGo K-1 cells, always higher than their expression in non-cancer nasal epithelial cells in culture. An obvious difference was the unimodal expression of DAF in the lung cancer cell lines that was in contrast to the bimodal expression of DAF in the non-cancer cells (Fig. 1d). In the non-cancer cells there were two cell subpopulations in terms of DAF expression. The major one expressed less DAF (Fig. 1d). In contrast, in the lung cancer cell lines there was only one, uniform cell population that exhibited a higher DAF expression. The significance of these differences in DAF expression is not clear, but high DAF expression may affect cancer cell susceptibility to NK cells [9] and DAF might be involved in transmembrane signalling [15].
Several reasons could theoretically account for the differences we found in cell membrane CIP expression. One reason is the constitutive difference of malignant versus non-malignant nature of the studied cells. A second reason might be the difference in the anatomical origin along the respiratory tract, bronchial versus nasal, although in a previous semiquantitative study we were not impressed by anatomical differences in CIP expression between bronchial and nasal epithelium [3]. A third reason might be the difference between types of cell culture systems, cell lines (cancer cells) and a primary cell culture (nasal cells). Of these three reasons, the first explanation, that the differences in expression resulted from malignancy, is particularly supported by other investigators, as well as by our own observations in vivo. Altered expression of cell membrane CIP has been described in various malignancies, although human lung cancer has scarcely been investigated. Increased levels of MCP, DAF and CD59 were described in haematological and in solid malignancies [7–9], and a change in antigenic determinant of DAF was suggested to occur in human lung cancer cell lines [16]. The present in vitro observation of increased cell membrane CIP expression in human lung cancer cell lines is also in accordance with our previous in vivo observation that CIP is over-expressed in human lung cancer tissue [3]. It might well be that lung cancer cell membrane expression in vivo is even further up-regulated by tissue cytokines and growth factors that promote lung cancer growth and cancer escape from the host's immunosurveillance.
The human lung cancer cell lines that we studied were extremely resistant to complement-mediated lysis (Figs 2 and 3). To the best of our knowledge, lung cancer resistance to complement-mediated lysis was not previously studied, but resistance of some other solid tumours to complement was found to be high and CIP molecules were suggested as an escape mechanism of tumour cells from the host's immunosurveillance system [7–9].
The lung cancer cell lines that we studied were not lysed by complement, because complement activation upon the cell membrane of these cells has been limited in its early steps (Figs 5 and 6). Spontaneous adsorption of complement C3-related fragments upon lung cancer cells was very low. Increasing NHS concentration had no effect on spontaneous C3 adsorption upon NCI-H596 cells, and in ChaGo K-1 cells only 50% of NHS resulted in increased C3 adsorption (Fig. 5). These findings are in sharp contrast to the pattern of spontaneous adsorption of C3-related fragments upon non-cancer human nasal respiratory epithelium [4]. Even the formation of cell membrane immunocomplexes, by using anti-NEC and anti-CEA antibodies, in the presence of anti-MCP and anti-DAF or the addition of LPS to NHS, did not result in a significant increase in adsorption of C3-related fragments upon the lung cancer cells (Fig. 6). This coincides with our results that such an approach is insufficient for inducing complement-mediated lysis of these resistant cancer cell lines (Figs 2 and 3). In contrast, formation of these cell membrane immunocomplexes upon non-cancer human nasal respiratory epithelium, or addition of the same type of LPS to NHS, resulted in significant complement-mediated lysis of these non-cancer cells [5].
Although we showed that complement is poorly activated upon the cell membrane of these lung cancer cell lines, the exact mechanism of increased resistance by these cells to complement-mediated lysis is not clear. An apparent discrepancy exists between the functional significance of DAF and CD59 expressed by the lung cancer cell lines, as suggested by the results of PIPLC treatment, on the one hand, and the inability of neutralizing antibodies against DAF and CD59 to increase both cell lysis and cell membrane complement activation on these cells, on the other (Figs 4 and 7). These discrepant findings are outstanding, were not described before, and might be explained in one or more ways. It might be that PIPLC affects the cell membrane attachment of not only DAF and CD59, but also of other as yet unidentified lung cancer cell line cell membrane complement inhibitory proteins, that are GPI-anchored, as are DAF and CD59. Alternatively, it is possible that DAF and CD59 operate synergistically with additional, unknown cell membrane complement regulator(s) on the lung cancer cell lines. According to this possibility, when PIPLC detaches DAF and CD59, this synergistic mechanism is severely damaged and allows activation of complement upon the cell membrane. In this regard, it is worth mentioning that an additional cell membrane complement inhibitory protein of a different type was described recently in malignant human melanoma cells [17]. Another, less likely possibility might be that the active site of DAF and CD59 is altered in these lung cancer cell lines so that neutralizing antibodies against normal CIP molecules can still immunologically recognize these regulators upon lung cancer cells, but are no longer capable of inactivating them. Under-saturation of lung cancer cell line CIP molecules, by these neutralizing antibodies, does not seem to be the reason for the inability to neutralize them, since we used saturating dilutions of antibodies, as judged by flow cytometry.
In conclusion, in this study we showed that human lung cancer cell lines ChaGo K-1 and NCI-H596 express cell membrane CIP molecules MCP, DAF and CD59 as do human lung cancer cells in vivo, but that the level of this expression in vitro is essentially higher than the level of cell membrane CIP expression in cell cultures of normal respiratory epithelium. In addition, we showed that the studied lung cancer cell lines were much more resistant to complement-mediated lysis than were normal cells. The high resistance to complement exhibited by ChaGo K-1 and NCI-H596 human lung cancer cell lines was due to the inability of serum complement to be activated upon these cells. This inability to activate complement seems to be strongly dependent on the presence of GPI-anchored cell membrane complement inhibitors. Whether these GPI-anchored molelcules are DAF and CD59, or perhaps, as yet unidentified cell membrane complement inhibitors, unique to lung cancer cells, awaits further research. These results suggest that the biology of cell membrane CIP molecules in human lung cancer differs from that of normal human respiratory epithelium and that human lung cancer might be especially resistant to complement-mediated lysis. Our in vitro model will, it is hoped, contribute to further understanding of human lung cancer biology and escape mechanisms.
Acknowledgments
This research was supported by the Israel Cancer Association with a grant donated by Ms Fanni M. Shor in memory of her late husband Mouritz, by the M. Modan Cancer Research Fund, Tel-Aviv University and by a contribution from Leah and Ariel Shenker. The authors thank Ms Sarah Gon-Paz for word processing and secretarial assistance.
REFERENCES
- 1.Muller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57:321–47. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]
- 2.Lachmann PJ. The control of homologous lysis. Immunol Today. 1991;12:312–5. doi: 10.1016/0167-5699(91)90005-E. [DOI] [PubMed] [Google Scholar]
- 3.Varsano S, Frolkis I, Ophir D. Expression and distribution of cell-membrane complement regulatory glycoproteins along the human respiratory tract. Am J Respir Crit Care Med. 1995;152:1087–93. doi: 10.1164/ajrccm.152.3.7545058. [DOI] [PubMed] [Google Scholar]
- 4.Varsano S, Frolkis I, Shapiro H, Ophir D. Human nasal epithelium adsorbs complement C3-related fragments and expresses cell-membrane complement regulatory proteins. Laryngoscope. 1996;106:599–604. doi: 10.1097/00005537-199605000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Varsano S, Frolkis I, Rashkovsky L, Ophir D, Fishelson Z. Protection of human nasal respiratory epithelium from complement mediated lysis by cell-membrane regulators of complement activation. Am J Resp Cell Mol Biol. 1996;15:731–7. doi: 10.1165/ajrcmb.15.6.8969267. [DOI] [PubMed] [Google Scholar]
- 6.Reiter Y, Fishelson Z. Targeting of complement to tumor cells by heteroconjugates composed of antibodies and of the complement component C3b. J Immunol. 1989;142:2771–7. [PubMed] [Google Scholar]
- 7.Hakulinen J, Meri S. Expression and function of the complement membrane attack complex inhibitor protectin (CD59) on human breast cancer cells. Lab Invest. 1994;71:820–7. [PubMed] [Google Scholar]
- 8.Brasoveanu LI, Altomonte M, Fonsalti E, et al. Levels of cell membrane CD59 regulate the extent of complement mediated lysis of human melanoma cells. Lab Invest. 1996;74:33–42. [PubMed] [Google Scholar]
- 9.Fiberg RW, White W, Nicholson-Weller A. Decay accelerating factor expression on either effector or target cells inhibits cytotoxicity by human natural killer cells. J Immunol. 1992;149:2055–60. [PubMed] [Google Scholar]
- 10.Cho SW, Oglesby TJ, Hsi BL, Adams EM, Atkinson JP. Characterization of three monoclonal antibodies to membrane cofactor protein (MCP) of the complement system and quantification of MCP by radioassay. Clin Exp Immunol. 1991;83:257–61. doi: 10.1111/j.1365-2249.1991.tb05624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinoshita T, Medof ME, Silber R, Nussenzweig V. Distribution of DAF in peripheral blood of normal individuals and patients with paroxysmal nocturnal hemoglobinuria. J Exp Med. 1985;162:75–92. doi: 10.1084/jem.162.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies A, Simmons DL, Hale G, Harrison RA, Tighe H, Lachmann PJ, Waldmann H. CD59, an LY-6-like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex on homologous cells. J Exp Med. 1989;170:637–54. doi: 10.1084/jem.170.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henner DJ, Yang M, Chen E, Hellmiss R, Rodriguez H, Low MG. Sequence of the Bacillus thuringiensis phosphatidyl inositol specific phospholipase C. Nucl Acids Res. 1988;16:10383. doi: 10.1093/nar/16.21.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Low MG, Striernberg J, Waneck GL, Flavell RA, Kincade PW. Cell-specific heterogeneity in sensitivity of phosphatidylinositol anchord membrane antigens to release by phospholipase C. J Immunol Methods. 1988;113:101–11. doi: 10.1016/0022-1759(88)90386-9. [DOI] [PubMed] [Google Scholar]
- 15.Shenoy-Scaria AM, Kwong J, Tujita T, Olszowy MW, Shaw AS, Lublin DM. Signal transduction through decay-accelerating factor: interaction of glycosyl-phosphatidylinositol anchor and protein tyrosine kinases p561ck and p59fynl. J Immunol. 1992;149:3535–41. [PubMed] [Google Scholar]
- 16.Sakuma T, Kodama K, Hara T, Eshita Y, Shibata N, Matsumoto M, Seya T. Levels of complement regulatory molecules in lung cancer: disappearance of the D17 epitope of CD55 in small-cell carcinoma. Jpn J Cancer Res. 1993;84:753–9. doi: 10.1111/j.1349-7006.1993.tb02040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ollert MW, Kadlec JV, Petrella EC, Bredehorst R, Voge1 CW. Molecular basis of complement resistance of human melanoma cells expressing the C3-cleaving membrane protease p65. Cancer Res. 1993;53:592–9. [PubMed] [Google Scholar]