

Abstract
Recombinant human deoxyribonuclease I (rhDNase) may be an effective therapeutic for the treatment of systemic lupus erythematosus (SLE). The pharmacodynamics of rhDNase in serum was investigated using two activity assays: one based on hydrolysis of a radiolabelled phage DNA and the other based on hydrolysis of human chromatin. The concentration of endogenous immunoreactive DNase in sera from 16 normal subjects was 3.2 ± 1.4 ng/ml (mean ± s.d.); however, low levels or no nuclease activity were detected in the same sera, suggesting the presence of DNase inhibitors. We assessed the ability of rhDNase to degrade DNA in undiluted serum, since the observed inhibition of endogenous DNase was reversed upon dilution. Addition of rhDNase to undiluted serum at a concentration of 50–100 ng/ml was necessary for degradation of radiolabelled phage DNA. The activity of rhDNase added to serum from normal subjects and SLE patients was similar. rhDNase degraded human chromatin and chromatin/anti-DNA immune complexes in serum with similar potency (EC50 ≈ 100–200 ng/ml). A 500-fold variation in the chromatin/anti-DNA stoichiometry did not significantly affect the digestion of these immune complexes by rhDNase in buffer. These results indicate that a minimum rhDNase concentration of 50–100 ng/ml in serum was required to achieve detectable catalytic activity and that the presence of antibodies to DNA did not inhibit the degradation of DNA/anti-DNA immune complexes.
Keywords: systemic lupus erythematosus, DNA, chromatin, DNase I, immune complexes, enzyme assays
INTRODUCTION
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by deposition of antinuclear autoimmune antibodies in a variety of tissues. Complement fixation at the sites of deposition causes vasculitis and chronic inflammatory damage to the affected organ systems, including the kidney, skin, joints, and central nervous system. The most common cause of death in untreated SLE is renal failure due to repeated exacerbations of inflammation. Flares in SLE activity are controllable with aggressive doses of corticosteroids or cytotoxic agents such as cyclophosphamide [1], but long-term immunosuppression is responsible for substantial morbidity and mortality.
Numerous reports implicate anti-chromatin antibodies, in particular anti-double-stranded DNA (anti-dsDNA) antibodies, as a principal causative factor in SLE [2,3]. However, the pathogenicity of antibodies to DNA versus other antigens and the mechanism responsible for the initiation of the autoimmune response in lupus are less clear. Flares of disease activity are correlated with prior increases in circulating levels of anti-dsDNA antibodies [4], especially in patients with SLE nephritis. There is evidence that anti-DNA antibodies are selectively retained in the kidneys [5] and immunofluorescence studies show that anti-DNA is bound to the glomeruli of SLE patients [6]. Deposition of immune complexes in the kidney is highly correlated with progressive renal dysfunction in SLE patients [7,8]. It is unclear if this binding is due to deposition of circulating immune complexes [9] or to direct binding of anti-DNA to cross-reactive glomerular antigens [10].
Several lines of evidence indicate that the immune response to DNA in SLE is antigen-driven [11–13]. For this reason, treatment of SLE with bovine DNase I was first attempted in the 1960s [14]. However, bovine DNase I was immunogenic in humans and chronic treatment was not possible. Recently, Macanovic et al. demonstrated that systemic adminstration of recombinant murine DNase I delayed the progression of SLE in NZB/W mice [15]. Therefore, systemic administration of Pulmozyme® recombinant human DNase I (rhDNase) may be an effective, non-immunogenic therapeutic for human SLE, as proposed by Lachmann [16]. rhDNase may hydrolyse the DNA component of membrane-deposited DNA/anti-DNA immune complexes, facilitating excretion of the anti-DNA antibodies into the urine and reducing glomerular inflammation. Alternatively, rhDNase may hydrolyse free and/or antibody-complexed DNA in blood, reducing the antigen load and thereby leading to a reduction in anti-DNA levels and in the deposition of immune complexes.
Most existing assays for rhDNase activity are not well suited to analysis of undiluted serum. Serum contains macromolecules that interfere with assays based on ultraviolet absorbance (the hyperchromicity assay [17]), visible absorbance (DNA-methyl green complexes [18]) or fluorescent dyes [19]. Reduction of these high background readings requires substantial dilution of the serum, resulting in an assay environment that no longer resembles the physiological milieu.
We have developed two assays for determination of DNase activity in undiluted human serum. The first assay, referred to as the 33P-DNA assay, uses purified 33P-labelled M13 phage DNA added to undiluted human serum. After digestion, the sample is precipitated with trichloroacetic acid (TCA) and the amount of TCA-soluble radioactivity is quantified. This assay provides a sensitive method of quantifying rhDNase activity in a sample that has a serum composition of 90%. To simulate the immune complexes that are present in human SLE, we developed a second assay that utilizes a substrate of human chromatin combined with anti-DNA IgG purified from SLE patients. These in vitro chromatin/anti-DNA immune complexes are added in small volumes to serum, resulting in a sample with a serum composition of 80–85%. Substrate digestion is monitored by agarose gels or by ELISA. Using these assays we investigated the ability of rhDNase to hydrolyse DNA and DNA/anti-DNA immune complexes in human serum.
MATERIALS AND METHODS
ELISA for DNase
DNase concentration in serum was measured by a two-site enzyme-linked immunoassay, using rhDNase I (Pulmozyme; Genentech, South San Francisco, CA) as the standard. The polyclonal antibodies used in the assay were generated in rabbits and were affinity-purified by standard procedures. Microtitre plates were coated overnight at 4°C with 100 μl/well of 62.5 ng/ml anti-rhDNase in 0.05 m sodium carbonate buffer pH 9.6. Between each of the following incubations, plates were washed with PBS/0.05% polysorbate 20. Non-specific binding sites were blocked by incubation for 1 h with 200 μl of ELISA buffer (25 mm HEPES–NaOH, 4 mm CaCl2, 4 mm MgCl2, 0.1% bovine serum albumin (BSA), 0.05% polysorbate 20, 0.03% proclin 300, pH 7.5). Diluted standards and samples (100 μl) were added to the wells and plates were incubated for 2 h, followed by addition of 100 μl of biotinylated anti-rhDNase (12.5 ng/ml) for 2 h. Streptavidin–β-galactosidase (100 μl; Boehringer-Mannheim, Indianapolis, IN), diluted 1:80 000, was added to each well. After incubation for 1 h, 50 μl of 1 mm 4-methylumbelliferyl-β-d-galactopyranoside (Molecular Probes, Eugene, OR) in substrate buffer (0.1 m sodium phosphate, 1 mm MgCl2, pH 7.3) were incubated in the wells in the dark for 24 h. The reaction was stopped by the addition of 200 μl of 0.15 m glycine pH 10.5, and fluorescence was read at an excitation wavelength of 360 nm and an emission wavelength of 460 nm. Sample concentration was interpolated from rhDNase standard curves (5–320 pg/ml) fitted to a four-parameter logistic equation [20]. A minimum 100-fold dilution of serum samples in ELISA buffer was required for accurate quantification; therefore, the detection limit for DNase in serum was 0.5 ng/ml.
33P-DNA assay for DNase activity
This assay measures the enzymatic degradation of purified DNA in serum and various buffers. Single-stranded M13 DNA template (Gibco-BRL, Gaithersburg, MD) was used to direct the synthesis of a 33P-labelled complementary DNA strand. This strand was synthesized using a 23-base primer directed at the polylinker region, α-33P-dATP (Amersham, Arlington Heights, IL), unlabelled dNTPs and the Klenow fragment of DNA polymerase I. Double-stranded 33P-M13 DNA was isolated from unincorporated nucleotides by passage through Sephadex G-50 spin columns (Boehringer-Mannheim). The 33P-M13 DNA was made up to a constant specific radioactivity of 0.70 ± 0.08 μCi/μg by addition of salmon testes DNA (Sigma Chemical Co., St Louis, MO), resulting in a final total DNA concentration of 81 μg/ml. 33P-DNA mixture (5 μl) was added to 95 μl of serum/buffer samples in the wells of microtitre plates. The contents of the wells were mixed by rotary agitation of the plates and were incubated at room temperature or at 37°C for 2 h. DNase activity was stopped by sequential addition of 100 μl ice-cold 50 mm EDTA and 100 μl ice-cold 20% TCA, followed by mixing. The microtitre plates were centrifuged at 1230 g for 15 min at 4°C and 50 μl of the supernatant were collected for determination of acid-soluble radioactivity in a scintillation counter. The percentage of solubilized radioactivity was linearly proportional to incubation time up to 2 h and was linearly proportional to DNase concentrations that solubilized ≤ 40% of the added radioactivity. Samples that solubilized > 40% of the added radioactivity were reassayed with a shorter incubation period. Reaction velocity was expressed as the μg of 33P-DNA solubilized. min−1. ml−1. The noise level of the method, determined by analysing 16 sera that were treated with 10 mm EDTA to inhibit any endogenous DNase, had a mean value of 8.5 × 10−5μg 33P-DNA solubilized. min−1. ml−1. The limit of detection was defined as 10 times the noise level.
Isolation of human anti-DNA IgG fraction from SLE patient sera
Serum samples from SLE patients were assayed for anti-DNA concentrations in the Farr radioimmunoassay [21] at Specialty Laboratories (Santa Monica, CA). Sera with high titres of anti-DNA antibodies were pooled. The IgG fraction was isolated using ammonium sulfate precipitation and anion exchange chromatography over a DE-52 column. Purity was > 95% by SDS–PAGE under reducing and non-reducing conditions. The total protein concentration was 13.3 mg/ml, based on an extinction coefficient of 1.5 at 280 nm, and the anti-DNA concentration was 1800 U/ml by the Farr assay.
Isolation of human chromatin from buffy coats
Buffy coat fractions (Peninsula Blood Bank, Burlingame, CA) from six units of human blood were pooled and leucocytes were isolated using multiple rounds of hypotonic lysis. Crude human chromatin was isolated from the leucocytes using a minor modification of the detergent lysis protocol of Trask & VanDenEngh [22]. The chromatin pellets were resuspended in 50 mm HEPES–KOH pH 8.0/5 mm MgSO4/0.1 mm disodium EDTA/1 mm dithiothreitol/40% glycerol (w/v). DNA concentration was quantified using the diaminobenzoic acid method [23] with salmon testes DNA as a standard.
In vitro chromatin/anti-DNA immune complexes
Chromatin, at a final concentration of 120 μg/ml DNA, was incubated with human anti-DNA IgG fraction at a final concentration of 180 U/ml for 3 h at 37°C in PBS or serum. For certain experiments, different stoichiometries of chromatin and human anti-DNA IgG were used as described in the figure legends. Immune complexes were then stored at 4°C and removed for assay as needed. Formation of immune complexes was verified by the chromatin/immune complex ELISA described below.
Digestion of chromatin and chromatin/anti-DNA immune complexes with rhDNase
Initial stock solutions of rhDNase were diluted in ELISA buffer. Typically, 80 μl of serum or PBS, 10 μl of chromatin or immune complexes and 10 μl of rhDNase were incubated in a 37°C water bath for 2 h. Digestion was stopped by addition of EDTA to 25 mm. Samples were divided into two aliquots. One aliquot was used for immune complex analysis by ELISA. The other aliquot was prepared for gel analysis by addition of 2% SDS and 1.0 mg/ml Proteinase K solution (Boehringer-Mannheim), followed by overnight digestion at 50°C. Samples were analysed by electrophoresis on 2.7% MetaPhor agarose (FMC, Rockland, ME)/0.2% SDS gels in TBE buffer, followed by post-staining with 1.0 μg/ml ethidium bromide. Gels were scanned with the FotoAnalyst system (Fotodyne, Hartland, WI), and bands were integrated using Collage software (Fotodyne).
Chromatin/immune complex ELISA
The volumes used in the following procedure were 100 μl/well unless noted otherwise. Microtitre plates were coated with 1.0 μg/ml of anti-histone MoAb (Boehringer-Mannheim) in 50 mm carbonate buffer pH 9.6. The plate was washed three times with PBS/0.05% polysorbate 20. Non-specific protein adsorption was then blocked with 200 μl/well of PBSGT buffer (PBS/0.1% gelatin/0.05% polysorbate 20/0.03% proclin 300) and the plate was incubated for 1–2 h. Immune complexes digested with rhDNase were diluted 1:50 into PBSGT followed by serial 1:3 dilutions. After plate washing as above, the diluted samples were added and incubated for 2 h. The plate was washed and a 1:5000 dilution of peroxidase-conjugated goat anti-human IgG, Fc-specific (Pierce, Rockford, IL) was added, followed by incubation for 1.5–2 h. After washing, colour was developed with 0.4 mg/ml o-phenylenediamine in PBS/0.03% hydrogen peroxide. Development was stopped with 4.5 n sulfuric acid, and the absorbance at 492 nm was measured using a reference wavelength of 405 nm.
RESULTS
Serum contains inhibitors of DNase
The concentration of endogenous DNase in sera from 16 normal subjects, measured by ELISA, was 3.2 ± 1.4 ng/ml (mean ± s.d.). However, analysis of these sera in the 33P-DNA assay indicated that most of the endogenous DNase was inactive. Although other nucleases in addition to DNase I may be present in human serum, low levels of nuclease activity were detected in only four of the 16 undiluted sera (limit of detection = 8.5 × 10−4). The mean nuclease activity in these four sera was 1.1 × 10−3 μg DNA solubilized. min−1. ml−1. In contrast, the activity of 3.2 ng/ml rhDNase under the same assay conditions in a hypotonic buffer was 2.0 × 10−2 μg DNA solubilized. min−1. ml−1. The composition of the hypotonic buffer used in these experiments was based on earlier data indicating that these conditions of pH, ionic composition and tonicity were near optimal for rhDNase activity [18]. These data suggested the presence of DNase inhibitors in serum.
The relative potency of rhDNase added to various media was investigated using the 33P-DNA assay (Fig. 1). In hypotonic buffer, the EC50 for hydrolysis of 33P-DNA by rhDNase was 1.8 ng/ml under these assay conditions. Addition of physiological concentrations of sodium chloride (150 mm NaCl) to the hypotonic buffer inhibited rhDNase activity, resulting in a shift of the dose–response curve to the right (EC50 = 17 ng/ml), in agreement with recently published data [24]. Addition of rhDNase to 33P-DNA in undiluted human serum shifted the dose–response curve even further to the right (EC50 = 160 ng/ml). Thus, human serum contains factor(s) in addition to NaCl that inhibit rhDNase.
Fig. 1.

Dose–response of rhDNase added to various media. The concentration of rhDNase added to hypotonic buffer (•; 25 mm HEPES, 2.5 mm CaCl2, 1 mm MgCl2, pH 7.5), isotonic buffer (▪; same as hypotonic buffer but containing additional 150 mm NaCl), and undiluted serum (▾) is plotted on the abscissa. DNase activity was measured by the 33P-DNA assay as described in Materials and Methods following incubation for 2 h at 37°C.
Reversibility of the inhibition produced by serum factors was investigated in a second experiment. Human serum was diluted in hypotonic buffer prior to the addition of 33P-DNA. As in the first experiment, when 33P-DNA was added to undiluted serum, there was no detectable hydrolysis of the substrate. However, detectable activity of endogenous serum nucleases was observed after a two-fold or greater dilution of the serum (data not shown). Similar results were obtained when rhDNase was added to serum at a final concentration of 100 ng/ml prior to dilution and addition of the 33P-DNA substrate. Therefore, to obtain data relevant to conditions in vivo, it is important to assay DNase activity by a method that does not require significant dilution of serum samples.
Minimum rhDNase concentration required to degrade purified DNA in serum
To determine the minimum active concentration of rhDNase in serum, dose–response curves were determined in serum obtained from normal subjects and from SLE patients. In these experiments, varying concentrations of rhDNase were added to undiluted serum samples prior to addition of 33P-DNA substrate (Fig. 2). Addition of 50–100 ng/ml of rhDNase to serum from normal subjects was required to produce detectable nuclease activity under these assay conditions. The EC50 of rhDNase added to this pool of normal serum was 670 ng/ml. The somewhat lower EC50 determined previously (Fig. 1) reflects the variation typically observed in different serum pools.
Fig. 2.
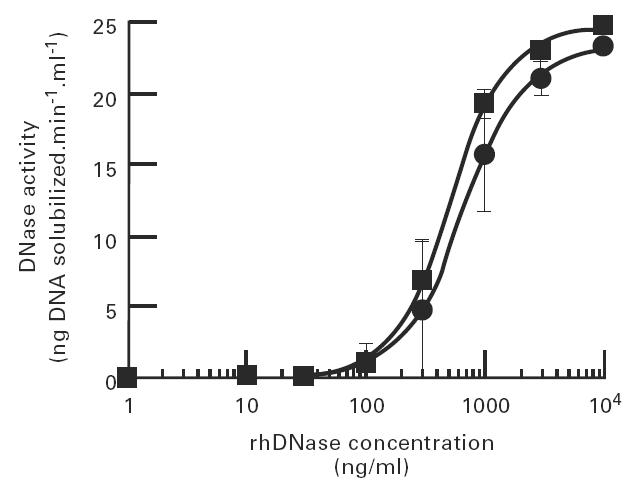
Dose–response of rhDNase added to normal and systemic lupus erythematosus (SLE) sera. rhDNase and 33P-DNA were added to normal sera (•, n = 6) and SLE sera (▪, n = 4) with minimal dilution of the samples. DNase activity was measured as described in Materials and Methods following incubation at 37°C for 2 h.
The dose–response curves obtained with sera from four SLE patients and six normal subjects were similar (EC50 = 500 ng/ml; Fig. 2). The activity of rhDNase added to an addtional 24 of 25 individual SLE sera was not statistically different from the activity measured in normal sera (data not shown). However, DNase activity was markedly decreased in serum from one SLE patient with a high titre of anti-DNA antibodies. The activity of rhDNase was not correlated with the titre of antibodies to DNA measured in the other 24 SLE sera. Thus, in most SLE sera examined, the presence of anti-DNA antibodies did not inhibit degradation of the 33P-DNA substrate. However, it is possible that the antibodies in these sera did not bind to purified 33P-DNA.
Formation of in vitro chromatin/anti-DNA immune complexes
We sought to compare rhDNase digestion of chromatin/anti-DNA immune complexes and of chromatin alone. An IgG fraction was isolated from pooled sera from several SLE patients exhibiting high anti-DNA titres in the Farr assay [21]. We isolated large quantities of human chromatin from buffy coat (leucocyte-rich layers of centrifuged anti-coagulated blood) specimens from a regional blood bank. The isolated chromatin was analysed by micrococcal nuclease digestion and agarose gel electrophoresis, which confirmed the presence of polynucleosomal DNA characteristic of that present in immune complexes isolated from SLE patients (data not shown). To create chromatin/anti-DNA immune complexes in vitro, this anti-DNA IgG fraction (final concentration 180 U/ml) was incubated with the human chromatin preparation. The formation of chromatin/IgG immune complexes was assayed by ELISA (Fig. 3). Chromatin alone did not produce a signal in this assay. Incubation of anti-DNA IgG with chromatin, however, produced a signal in the assay that was decreased by digestion with rhDNase in a dose-dependent manner, indicating the formation of chromatin/anti-DNA immune complexes. Chromatin added to a pool of SLE sera also produced a signal in this assay which was reduced by digestion with rhDNase, suggesting the formation of immune complexes with endogenous anti-DNA. The highest signal was attained when chromatin/anti-DNA complexes were added to the SLE serum, probably due to the formation of immune complexes composed of both exogenous and endogenous anti-DNA.
Fig. 3.

Characterization of in vitro chromatin/anti-DNA IgG immune complexes. Human chromatin (120 μg/ml) plus anti-DNA IgG (180 U/ml by Farr assay), or chromatin alone, were added to PBS or to pooled systemic lupus erythematosus (SLE) serum and incubated for 3 h at 37°C to allow immune complex formation. Aliquots of these samples were then digested with increasing concentrations of rhDNase (10 ng/ml up to 10 μg/ml) for 2 h at 37°C. DNase digestion was stopped by addition of EDTA to 25 mm. Samples were assayed at a 1:150 dilution in the chromatin/immune complex ELISA as described in Materials and Methods. Results are plotted as ELISA absorbance on the ordinate versus rhDNase concentration on the abscissa. Curves are identified on the plot.
Further evidence that co-incubation of chromatin and anti-DNA IgG produced immune complexes was obtained by fractionating samples over a Superose 12 gel filtration column. Fractions were collected and assayed in the chromatin immune complex ELISA. The observed ELISA reactivity in the chromatin/anti-DNA sample was in the high molecular weight (> 670 kD) fractions (data not shown). Anti-DNA IgG alone and chromatin alone were unreactive in this ELISA. These results are consistent with the formation of chromatin/anti-DNA complexes.
Digestion of chromatin in PBS and serum by rhDNase
Digestion of chromatin in PBS and serum by rhDNase was evaluated by agarose gel electrophoresis (Fig. 4). Significant digestion of chromatin in PBS was first observed at 30 ng/ml rhDNase, with extensive digestion at 100 ng/ml rhDNase (Fig. 4a). Concentrations of rhDNase > 300 ng/ml completely digested the chromatin. In human serum, higher concentrations of rhDNase were necessary to digest the chromatin. Significant digestion was first observed at 100 ng/ml rhDNase and complete degradation was observed at ≥ 3 μg/ml rhDNase (Fig. 4b). Thus, as observed in the 33P-DNA assay (Fig. 1), the catalytic activity of rhDNase was inhibited by factors present in serum.
Fig. 4.

Digestion of human chromatin/anti-DNA IgG immune complexes and chromatin alone by rhDNase. Aliquots of chromatin alone in PBS (a), chromatin alone in systemic lupus erythematosus (SLE) serum (b), chromatin/anti-DNA ICs in PBS (c) and chromatin/anti-DNA ICs in SLE serum (d) were digested with increasing concentrations of rhDNase for 2 h at 37°C. The final chromatin concentration was 120 μg/ml DNA and the final anti-DNA IgG concentration was 180 U/ml by Farr assay. Digestion was stopped with EDTA, followed by overnight proteinase K digestion and agarose gel electrophoresis as described in Materials and Methods. The concentrations of DNase used, in ng/ml, are listed above each lane. Sizes of DNA size markers used, in base pairs (bp), are listed at the sides of each panel. (b,d) Undigestible ethidium-stained material at ∼1 kb has been observed in 11 individual SLE sera as well as pooled normal sera not containing added chromatin.
Digestion of chromatin/anti-DNA immune complexes in buffer and serum
Chromatin/anti-DNA immune complexes made in vitro were added to buffer or serum and were digested with rhDNase at 37°C for 2 h. Evaluation by agarose gel electrophoresis (Fig. 4c) revealed that 30 ng/ml rhDNase significantly digested the chromatin and that digestion was essentially complete at 300 ng/ml rhDNase. Thus, the digestion of chromatin/anti-DNA complexes in PBS by rhDNase (Fig. 4c) was similar to the digestion of chromatin alone (Fig. 4a). An agarose gel of chromatin/anti-DNA immune complexes in serum digested with rhDNase is shown in Fig. 4d. Significant digestion of chromatin/anti-DNA complexes was first observed at 100 ng/ml rhDNase, similar to chromatin alone (Fig. 4b). Complete digestion of complexes was observed at rhDNase concentrations > 1 μg/ml. Therefore, the dose–response profile for digestion of chromatin/anti-DNA complexes in serum was similar to that of chromatin alone.
To compare the above rhDNase dose–responses, we scanned the gels and integrated two regions of each lane: the area ≥ 500 bp (representing DNA sizes of at least trinucleosomes) and the area between 130 bp and 300 bp (representing mono- and dinucleosomal DNA). The ratio of high mol. wt DNA/low mol. wt DNA was calculated for each dose. The EC50 values estimated from these curves are shown in Table 1. The EC50 values for digestion of chromatin/anti-DNA complexes and chromatin alone were identical in PBS (15 ng/ml). In SLE serum, chromatin/anti-DNA complexes had an EC50 value (200 ng/ml) that was approximately twice that of chromatin alone (120 ng/ml). These data indicate that free chromatin and chromatin/anti-DNA complexes were degraded by rhDNase at similar rates. In summary, degradation of chromatin in serum required more rhDNase than degradation of chromatin in PBS, but the presence of anti-DNA complexed to the chromatin did not significantly affect chromatin hydrolysis in either matrix.
Table 1.
Estimated recombinant human deoxyribonuclease (rhDNase) concentrations for 50% digestion (EC50) of chromatin/anti-DNA immune complexes or chromatin alone in PBS and human serum

Digestion of chromatin/anti-DNA at different stoichiometries
The stoichiometry of our in vitro chromatin/anti-DNA immune complexes may be very different from those present in SLE patients. To determine whether the digestion of DNA was affected by the stoichiometry of the immune complexes, we compared the ability of rhDNase to degrade immune complexes made with different ratios of anti-DNA and chromatin. Degradation of DNA was measured by the chromatin/immune complex ELISA. Since the reactivity of immune complexes in this assay is dependent upon their stoichiometry, we normalized the data to the maximum response obtained at each ratio of chromatin to anti-DNA (Fig. 5). The EC50 values from six complexes that varied 500-fold in DNA/antibody ratio were between 2.9 ng/ml and 8.0 ng/ml. These data indicate that the degree of chromatin saturation by anti-DNA antibody, over an estimated stoichiometry of between 13 and 6570 DNA base pairs per antibody, did not influence the ability of DNase to digest the chromatin.
Fig. 5.
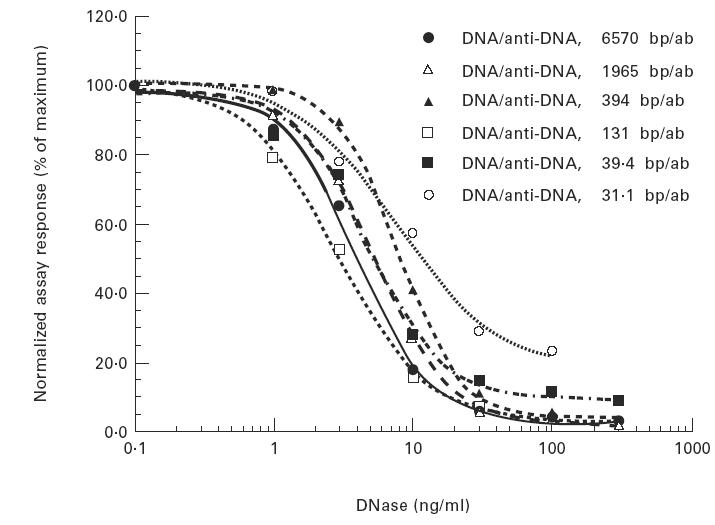
Digestion of human chromatin/anti-DNA IgG immune complexes at six different stoichiometries by rhDNase. Varying concentrations of chromatin and anti-DNA IgG were incubated in PBS for 3 h at 37°C, followed by digestion with increasing concentrations of rhDNase for 2 h at 37°C. Digestion was stopped with 25 mm EDTA. Aliquots were analysed in the chromatin/immune complex ELISA as described in Materials and Methods. Due to widely varying maximal absorbances at different stoichiometries, each dose–response curve was normalized to its maximum (no DNase) response. Results are plotted as percent of maximal response versus rhDNase concentration. Dose–response curves were drawn using a four-parameter data reduction algorithm within Kaleidagraph. The calculated EC50 are not shown in the figure, but were all between 3 and 8 ng/ml rhDNase. Although the shape of the 13 bp/ab curve looks different from the others, this is due only to the low ELISA signal obtained at this stoichiometry; the EC50 for this sample was 7.9 ng/ml.
DISCUSSION
The results of our in vitro studies indicate that normal physiological concentrations of endogenous DNase in human serum are insufficient to degrade DNA. Rat serum proteins have been shown to form complexes with rhDNase [25]. Actin and physiological saline are known to inhibit the activity of rhDNase [24,26,27]. Inhibition of endogenous DNase and exogenous rhDNase added to serum is readily reversed by dilution of the serum prior to measurement of enzymatic activity. The number and identity of inhibitors DNase in serum is currently being investigated.
A goal of our in vitro studies was to estimate the serum concentration of rhDNase required for hydrolysis of DNA as a guide to dosing of rhDNase in lupus patients. Therefore, it was necessary to develop assays for DNase activity that required minimal dilution of the serum. The two activity assays presented here fit this criterion, with sample compositions of at least 80% serum after DNA substrate addition. In the 33P-DNA assay, the minimum concentration of rhDNase required to degrade DNA in serum is 50–100 ng/ml; this is approximately 15–30-fold higher than the concentration of endogenous serum DNase in normal subjects.
The DNA present in immune complexes of SLE patients is likely to be derived from cellular turnover. DNA isolated from human serum is resolved by agarose gel electrophoresis as an oligonucleosomal ladder characteristic of apoptotic cell death [28]. In the present study, we demonstrate that rhDNase can readily digest the DNA component of human chromatin, including the DNA within the nucleosome core particle. The EC50 for degradation of human chromatin in serum was 120 ng rhDNase/ml. The observation that the EC50 determined by the chromatin assay is somewhat lower than that determined by the 33P-DNA assay (150–670 ng/ml) may be attributable to the difference in the substrates and the assay detection methods. Detection of enzymatic activity in the 33P-DNA assay requires extensive degradation of DNA to 20 bp or less. In contrast, limited hydrolysis of the chromatin substrate to fragments of 1–3 kilobases can be detected readily in both the chromatin/immune complex ELISA and agarose gel electrophoresis.
Our observation that rhDNase degraded chromatin and chromatin/anti-DNA immune complexes with approximately equal potency indicates that antibodies from SLE patients do not protect the DNA from hydrolysis. The EC50 of DNA digestion by rhDNase was nearly identical when chromatin and chromatin/anti-DNA immune complexes were incubated in serum (120–200 ng/ml) for 2 h at 37°C. Furthermore, when chromatin and chromatin/anti-DNA immune complexes were digested by rhDNase in PBS, the EC50 values were identical (15 ng/ml). Anti-DNA antibodies from SLE sera may be displaced by DNA-modifying enzymes, much as histones are displaced from nucleosomes during transcription by RNA polymerase [29,30]. Support for this conclusion comes from our observation that rhDNase was equipotent in hydrolysing 33P-DNA in normal human sera and in 23 of 24 sera from SLE patients with varying titres of anti-DNA antibodies. Additional support is provided by a recent study in which an immunoglobulin fraction isolated from SLE serum did not inhibit DNase activity on a DNA/agarose substrate [27].
Data from chromatin/anti-DNA complexes at six different stoichiometries indicate that digestion of the complexes by rhDNase was unaffected by a 500-fold change in the ratio of DNA base pairs/anti-DNA antibody, ranging from 6570 bp/antibody to 13.1 bp/antibody. In calculating these ratios, we estimated that our human anti-DNA IgG preparation from SLE patients contained 4.7% specific anti-DNA, based on Williams' measurements of serum anti-DNA and IgG levels in nine patients [31]. Emlen et al. found that bivalent, but not univalent, binding of anti-DNA IgG protected 35–50 base pairs of calf thymus DNA from DNase digestion [32]. We observed no significant protection of chromatin by anti-DNA binding in our system; however, it is possible that the immunoglobulin binding was univalent in our system. In addition, protection of 35–50 base pairs of DNA would not be resolved in our agarose gels or ELISA.
Our in vitro data support the proposed therapeutic use of rhDNase for the in vivo destruction of antigen in systemic lupus. Degradation of free DNA by rhDNase in SLE patients may suppress the production of anti-DNA antibodies. Moreover, degradation of the DNA component of immune complexes may reduce or eliminate the inflammation in tissues that underlies morbidity in SLE. Physiological concentrations of endogenous DNase are not sufficient to degrade DNA in human serum. The results of our in vitro studies predict that systemic administration at a dose sufficient to achieve a blood rhDNase concentration of > 50–100 ng/ml may be effective in the treatment of SLE. Actin-resistant and hyperactive variants of rhDNase that are potentially effective at substantially lower concentrations have now been synthesized [24,33].
Acknowledgments
The authors would like to thank Drs Steve Shak, Peter Lachmann, Eugen Koren, Clark Pan and Bob Lazarus for helpful discussions during this work.
References
- 1.Klippel JH, Decker JL. Systemic lupus erythematosus. In: Zvaifler NJ, editor. Internal medicine. Boston: Little, Brown & Co.; 1990. pp. 1739–46. [Google Scholar]
- 2.Lefkowith JB, Kiehl M, Rubenstein J, et al. Heterogeneity and clinical significance of glomerular-binding antibodies in SLE. J Clin Invest. 1996;98:1373–80. doi: 10.1172/JCI118924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lefkowith JB, Gilkeson GS. Nephritogenic autoantibodies in lupus: current concepts and continuing controversies. Arthritis Rheum. 1996;39:894–903. doi: 10.1002/art.1780390605. [DOI] [PubMed] [Google Scholar]
- 4.terBorg EJ, Horst G, Hummel EJ, et al. Measurement of increases in anti-double stranded DNA antibody levels as a predictor of disease exacerbation in SLE. Arthritis Rheum. 1990;33:634–42. doi: 10.1002/art.1780330505. [DOI] [PubMed] [Google Scholar]
- 5.Yamada A, Miyakawa Y, Kosaka K. Entrapment of anti-DNA antibodies in the kidney of patients with systemic lupus erythematosus. Kidney Int. 1982;22:671–6. doi: 10.1038/ki.1982.228. [DOI] [PubMed] [Google Scholar]
- 6.Koffler D, Agnello V, Kunkel HG. Polynucleotide immune complexes in serum and glomeruli of patients with systemic lupus erythematosus. Am J. Pathol. 1973;74:109–24. [PMC free article] [PubMed] [Google Scholar]
- 7.Sasaki T, Muryoi T, Hatakeyama A, et al. Circulating anti-DNA immune complexes in active lupus nephritis. Am J Med. 1991;91:355–62. doi: 10.1016/0002-9343(91)90152-n. [DOI] [PubMed] [Google Scholar]
- 8.Malide D, Londono I, Russo P, et al. Ultrastructural localization of DNA in immune deposits of human lupus nephritis. Am J Pathol. 1993;143:304–11. [PMC free article] [PubMed] [Google Scholar]
- 9.Kramers C, Hylkema MN, Vanbruggen MC, et al. Anti-nucleosome antibodies complexed to nucleosomal antigens show anti-DNA reactivity and bind to rat glomerular basement membranes in vivo. J Clin Invest. 1994;94:568–77. doi: 10.1172/JCI117371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raz E, Brezis M, Rosenmann E, et al. Anti-DNA antibodies bind directly to renal antigens and induce kidney dysfunction in the isolated perfused rat kidney. J Immunol. 1989;142:3076–82. [PubMed] [Google Scholar]
- 11.Klinman DM, Shirai A, Ishigatsubo Y, et al. Quantitation of IgM and IgG-secreting B cells in the peripheral blood of patients with SLE. Arthritis Rheum. 1991;34:1404–10. doi: 10.1002/art.1780341110. [DOI] [PubMed] [Google Scholar]
- 12.Fredriksen K, Osei A, Sundsfjord A, et al. Systemic lupus erythematosus-related anti-dsDNA antibodies are induced by polyomavirus BK in lupus prone (NZB/W) F1 hybrids, but not in normal mice. Eur J Immunol. 1994;24:66–70. doi: 10.1002/eji.1830240111. [DOI] [PubMed] [Google Scholar]
- 13.Gilkeson GS, Pippen AM, Pisetsky DS. Induction of cross-reactive anti-dsDNA antibodies in preautoimmune NZB/W mice by immunization with bacterial DNA. J Clin Invest. 1995;95:1398–402. doi: 10.1172/JCI117793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lachmann PJ. The Scientific Basis of Medicine Annual Reviews 1967. London: British Postgraduate Medical Federation, The Athlone Press, University of London; 1967. Allergic reactions, connective tissue and disease; pp. 35–58. [PubMed] [Google Scholar]
- 15.Macanovic M, Sinicropi D, Shak S, et al. The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin Exp Immunol. 1996;106:243–52. doi: 10.1046/j.1365-2249.1996.d01-839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lachmann PJ. The in vivo destruction of antigen—a tool for probing and modulating an autoimmune response. Clin Exp Immunol. 1996;106:187–9. doi: 10.1046/j.1365-2249.1996.d01-856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunitz M. Crystalline desoxyribonuclease I: isolation and general properties. J Gen Physiol. 1950;33:349–62. doi: 10.1085/jgp.33.4.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinicropi D, Baker DL, Prince WS, et al. Colorimetric determination of DNase I activity with a DNA-methyl green substrate. Anal Biochem. 1994;222:351–8. doi: 10.1006/abio.1994.1502. [DOI] [PubMed] [Google Scholar]
- 19.Morgan AR, Evans DH, Lee JS, et al. Ethidium fluorescence assay. Part II. Enzymatic studies and DNA–protein interactions. Nucelic Acids Res. 1979;7:571–97. doi: 10.1093/nar/7.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marquardt DW. An algorithm for least squares estimation of nonlinear parameters. J Soc Ind Appl Math. 1963;11:413–41. [Google Scholar]
- 21.Smeenk R, Hylkema M. Detection of antibodies to DNA: a technical assessment. Mol Bio Rep. 1992;17:71–79. doi: 10.1007/BF01006401. [DOI] [PubMed] [Google Scholar]
- 22.Trask B, Van Den Engh G. Chromosomal and nuclei isolation with the MgSO4 procedure. Methods Cell Biol. 1990;33:363–7. doi: 10.1016/s0091-679x(08)60539-7. [DOI] [PubMed] [Google Scholar]
- 23.Kissane JM, Robins E. The fluorometric measurement of deoxyribonucleic acid in animal tissues with special reference to the central nervous system. J Biol Chem. 1958;233:184–8. [PubMed] [Google Scholar]
- 24.Pan CQ, Lazarus RA. Engineering hyperactive variants of human deoxyribonuclease I by altering its functional mechanism. Biochemistry. 1997;36:6624–32. doi: 10.1021/bi962960x. [DOI] [PubMed] [Google Scholar]
- 25.Mohler M, Cook J, Lewis D, et al. Altered pharmacokinetics of recombinant human deoxyribonuclease in rats due to the presence of a binding protein. Drug Metab Dispos. 1993;21:71–75. [PubMed] [Google Scholar]
- 26.Lazarides E, Lindberg U. Actin is the naturally occurring inhibitor of deoxyribonuclease I. Proc Natl Acad Sci USA. 1974;71:4742–6. doi: 10.1073/pnas.71.12.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macanovic M, Lachmann PJ. Measurement of deoxyribonuclease I (DNase) in the serum and urine of systemic lupus erythematosus (SLE)-prone NZB/NZW mice by a new radial enzyme diffusion assay. Clin Exp Immunol. 1997;108:220–6. doi: 10.1046/j.1365-2249.1997.3571249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rumore PM, Steinman CR. Endogenous circulating DNA in systemic lupus erythematosus–occurrence as multimeric complexes bound to histone. J Clin Invest. 1990;86:69–74. doi: 10.1172/JCI114716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Studitsky VM, Clark DJ, Felsenfeld G. A histone octamer can step around a transcribing polymerase without leaving the template. Cell. 1994;76:371–82. doi: 10.1016/0092-8674(94)90343-3. [DOI] [PubMed] [Google Scholar]
- 30.Lewin B. Chromatin and gene expression: constant questions, but changing answers. Cell. 1994;79:397–406. doi: 10.1016/0092-8674(94)90249-6. [DOI] [PubMed] [Google Scholar]
- 31.Williams RC, Malone CC, Huffman GR, et al. Active systemic lupus erythematosus is associated with depletion of the natural generic anti-idiotype anti-F(ab′)2 system. J Rheumatol. 1995;22:1075–85. [PubMed] [Google Scholar]
- 32.Emlen W, Ansari R, Burdick G. DNA/anti-DNA immune complexes: antibody protection of a discrete DNA fragment from DNase digestion in vitro. Clin Invest. 1984;74:185–90. doi: 10.1172/JCI111400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulmer JS, Herzka A, Toy K, et al. Engineering actin-resistant human DNase I for treatment of cystic fibrosis. Proc Natl Acad Sci USA. 1996;93:8225–9. doi: 10.1073/pnas.93.16.8225. [DOI] [PMC free article] [PubMed] [Google Scholar]