

Abstract
Expression of DAF (CD55) is enhanced on colonic epithelial cells of patients with ulcerative colitis (UC), and stool DAF concentrations are increased in patients with active disease. Cytokines are known to modulate DAF expression in various human cells, and lesions of UC reveal altered profiles of cytokine production. In this study, we evaluate the effects of various cytokines, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, and interferon-gamma (IFN-γ), on the synthesis and kinetics of DAF protein in HT-29 human intestinal epithelial cells. Using flow cytometry and an ELISA, we found that HT-29 cells constitutively express DAF on the cell surface and spontaneously release DAF into the culture supernatant under standard culture conditions. When the culture supernatant was centrifuged at 100 000 g, nearly a half of DAF was precipitated, indicating that one half of the released DAF was present as a membrane-bound form and the other half as a soluble form. Analysis of the culture supernatant of biotin surface-labelled HT-29 cells suggested that the soluble form DAF was derived by secretion from within the cell or by cleavage from the cell surface. Among the cytokines, IL-4 markedly, and IL-1β moderately, enhanced the expression and the release of DAF. Actinomycin D, cycloheximide, and brefeldin A inhibited the increase in DAF release induced by IL-4 and IL-1β stimulation. These results suggest that DAF is released from intestinal epithelial cells in response to cytokine stimulation and that IL-4 and IL-1β are possible cytokines involved in DAF release into the colonic lumen of patients with UC.
Keywords: decay-accelerating factor, cytokine, IL-4, HT-29
INTRODUCTION
Ulcerative colitis (UC) is a chronic inflammatory disease affecting the large bowel, and altered regulation of cell-mediated and humoral factor-mediated immune responses against intestinal constituents have been suggested to play a role in the development of the disease [1]. Among systems which are humoral factor-mediated, the complement system is one of the major effector pathways. We have previously shown that the activated fragment (C3b) of the third component of complement (C3) as well as degraded fragments (iC3b/C3dg) of C3 are deposited in the affected mucosa of UC, suggesting that activation and degradation of complement occur in mucosal lesions of UC [2].
Autologous complement activation is regulated by complement regulatory proteins which act to protect host cells. One of these, DAF (CD55), is a membrane-bound glycoprotein which inhibits the formation and promotes the catabolism of C3 and C5 convertases [3]. We have previously shown that expression of DAF is enhanced on the apical surface of colonic epithelial cells in UC mucosa in relation to the severity of mucosal inflammation [4]. We have found also that stool DAF concentrations in active UC patients are increased [5]. Since cytokines such as IL-4 are known to enhance DAF expression in various human cells [6–9], and altered profiles of cytokine production can be observed with UC lesions [10,11], it is likely that the enhanced expression and release of DAF in UC mucosa is related to the change in the cytokine profile which occurs with this disease. In the present study, we evaluated the effects of various cytokines on the synthesis and kinetics of DAF protein in HT-29 human intestinal epithelial cells, and found that IL-4 strongly enhanced both the expression and the release of DAF from HT-29 cells.
MATERIALS AND METHODS
Cytokines and antibodies
Recombinant human IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, and interferon-gamma (IFN-γ) were obtained from Genzyme (Cambridge, MA). Human DAF was purified from pooled human erythrocyte stroma as described [12], and mouse MoAb (1C6, IgG1 isotype) and rabbit polyclonal antibody to DAF were prepared as described [12,13]. Horseradish peroxidase (HRP)-labelled goat F(ab′)2 anti-rabbit IgG was obtained from Tago Inc. (Burlingame, CA). FITC-conjugated F(ab′)2 fragments of rabbit anti-mouse IgG were purchased from Dako (Glostrup, Denmark).
Cell culture
A human colonic adenocarcinoma cell line, HT-29, was obtained from Dainippon Pharmaceutical Co. (Osaka, Japan) and maintained in McCoy's 5A medium modified with l-glutamine (Dainippon Pharmaceutical Co.) containing 10% fetal calf serum (FCS), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco BRL, Gaithersburg, MD) at 37°C in an atmosphere containing 5% CO2. Experiments were performed using HT-29 monolayers from three to six passages. Cell viability was assessed by trypan blue exclusion.
Stimulation of cells
HT-29 cells were seeded into tissue culture dishes (Becton Dickinson, Oxnard, CA) at a concentration of 1.0 × 105 cells/cm2 and cultured for 48 h with McCoy's 5A medium containing FCS and then overnight with FCS-free medium. After washing, cells were incubated with FCS-free medium containing various cytokines at concentrations of 1–100 ng/ml or 1–100 U/ml for 4–24 h. Medium in the absence of cytokines served as the control.
After the culture supernatant was collected, cells were detached with PBS containing 50 mm EDTA. DAF is known to be attached to the cell membrane through a glycosylphosphatidylinositol (GPI) anchor [14]. Therefore, to determine the amount of cell surface DAF, cells were incubated with PBS containing phosphatidylinositol-specific phospholipase C (PIPLC) isolated from Bacillus cereus (Sigma Chemical Co., St Louis, MO) for 30 min at 37°C. After the PBS/PIPLC solution was collected, cells were added to PBS containing 1% Nonidet P-40 (Sigma), 10 mm EDTA, and 1 mm PMSF (Sigma), placed on ice for 30 min and sonicated.
To study the form of DAF in the culture supernatant, large fragments of detached cells were removed by centrifugation at 15 000 g for 15 min, and its supernatant was further centrifuged at 100 000 g for 60 min at 4°C.
Determination of DAF protein
The amount of DAF in each sample was determined with an ELISA using 1C6 mouse monoclonal and rabbit polyclonal antibodies as previously described [13]. Briefly, samples were added to the wells of microtitre plates coated with 1C6 mouse MoAb and incubated. After washing, rabbit polyclonal anti-DAF antibody was added to the wells. After further incubation and washing, bound rabbit antibody was detected with HRP-labelled goat F(ab′)2 anti-rabbit IgG and 2,2′-amino-bis-3-ethylbenzo-thiazoline-6-sulphonic acid (Sigma) as substrate. Optical densities (OD) at 415 nm were measured on an automated ELISA plate reader. A calibration curve was obtained from several dilutions of known quantities of purified DAF, and the concentrations of DAF were calculated. The ELISA assay was sensitive to 0.2 ng/ml and accurate to 6 ng/ml [13].
Flow cytometry
HT-29 cells were detached with PBS containing 50 mm EDTA. After washing with PBS containing 1% bovine serum albumin (BSA; Sigma), the cells were incubated with 1C6 anti-DAF antibody on ice for 30 min, then labelled with FITC-conjugated anti-mouse IgG for 30 min. Cells treated with PIPLC (100 mU/ml) were also examined as described above. Mouse MoAb of the IgG1 subclass specific for an irrelevant antigen was used as a negative control. After washing, 1.0 × 104 cells were analysed using a FACScan (Becton Dickinson).
Immunoprecipitation and Western blot analysis
Biotinylation of the cell surface with sulfosuccinimidobiotin (Pierce, Rockford, IL) was performed as previously described [15]. After washing, the cell monolayer was incubated in culture medium for 24 h. The supernatant was then collected, concentrated and dialysed against PBS. After washing, cells were either solubilized with PBS containing 1% Nonidet P-40, 10 mm EDTA, and 1 mm PMSF or incubated with PBS containing PIPLC, and PBS/PIPLC solution was collected. These specimens were preabsorbed with Sepharose CL-4B (Pharmacia Fine Chemicals, Piscataway, NJ). Cyanogen bromide-activated Sepharose beads were coupled with 1C6 anti-DAF antibody, and the 1C6-labelled beads were mixed and incubated with the specimens overnight at 4°C with continuous rotation. After washing, immunoconjugates were eluted with Laemmli sample buffer [16], subjected to 7.5% SDS–PAGE under non-reducing conditions, and transferred to a nitrocellulose membrane (BioRad, Hercules, CA) using Trans-Blot (BioRad). The membrane was then incubated with blocking reagent (Amersham Life Sciences, Aylesbury, UK), and biotin-labelled bands were detected using HRP-labelled streptavidin and a chemiluminescence-based detection kit (Hyperfilm-ECL and ECL detection reagent; Amersham) according to the manufacturer's protocols.
Absorption with streptavidin beads
Biotinylated HT-29 cells were cultured for 24 h in medium with or without addition of cycloheximide (100 μm). The culture medium was centrifuged at 100 000 g for 60 min, and its supernatant was collected. One hundred millilitres of streptavidin-agarose beads (50% slurry in PBS) (Pierce) were added to 1 ml of each sample and incubated with continuous rotation at 4°C for 24 h. As control, biotinylated cells were treated with PBS containing PIPLC; PBS/PIPLC solution was collected and reacted with the agarose beads. After the beads were removed by brief centrifugation, the amount of DAF in each sample was measured by ELISA.
Statistical analysis
For statistical analysis, Student's t-test was used.
RESULTS
Properties of DAF on HT-29 cells
Flow cytometric analysis indicated that HT-29 cells constitutively expressed DAF on the cell surface (Fig. 1a). PIPLC treatment of the cells raised the concentration of DAF in the PIPLC solution and lowered the amount in the cell lysate. The effects of treatment plateaued at a PIPLC concentration of 100 mU/ml (Fig. 2). Treatment at this concentration abrogated about 70% of the surface labelling of HT-29 cells with anti-DAF antibody (Fig. 1b). The amount of the PIPLC-sensitive cell surface DAF obtained without stimulation was estimated to be 9.21 × 10−16 g/cell (7.92 × 103 molecules/cell).
Fig. 1.
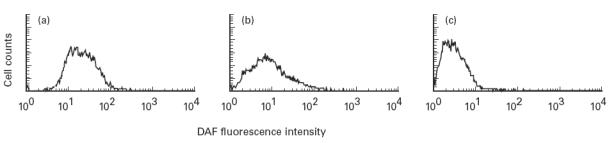
Cell surface expression of DAF on HT-29 cells and the effect of phosphatidylinositol-specific phospholipase C (PIPLC) treatment. Cells were detached, incubated with 1C6 anti-DAF antibody (a) or an irrelevant MoAb (c), then with an FITC-conjugated anti-mouse IgG, and analysed by flow cytometry. Cells treated with PIPLC (100 mU/ml) were also examined as described above (b). This experiment was repeated twice with similar results.
Fig. 2.
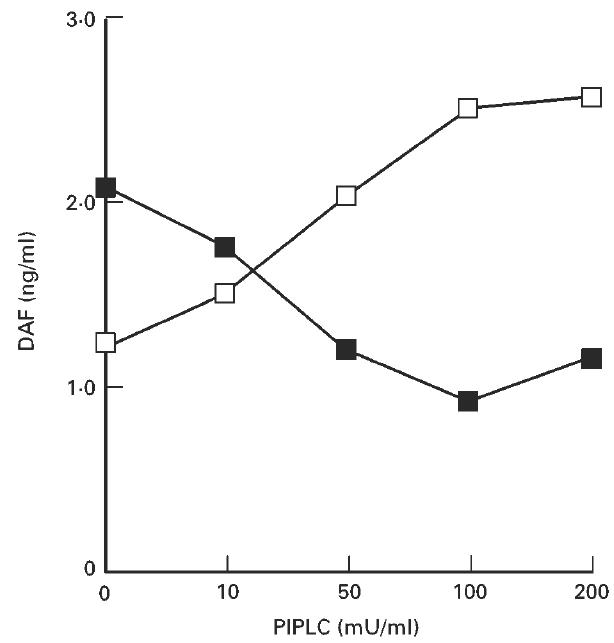
Release of DAF from the surface of HT-29 cells by phosphatidylinositol-specific phospholipase C (PIPLC) treatment. HT-29 cells (1.0 × 106 cells) were detached and treated with PIPLC. The amounts of DAF released into the PIPLC solution (□) and in the lysate of remaining cells (×100) (▪) were determined by ELISA. This experiment was repeated twice with similar results.
HT-29 cells spontaneously release DAF into the culture supernatant
As shown in Fig. 3a, HT-29 cells released DAF into the culture supernatant spontaneously and in a time-dependent manner under standard cell culture conditions. The mean amount of DAF released from HT-29 cells in 24 h was calculated to be 2.86 × 10−15 g/cell (2.46 × 104 molecules/cell). When cell surface proteins were labelled with biotin, biotinylated DAF was detected in the culture supernatant. The apparent molecular weight of the biotin-labelled DAF in the culture supernatant was almost the same as that of the membrane-bound form but slightly larger (about 3 kD) than that of PIPLC-released DAF (Fig. 3b).
Fig. 3.
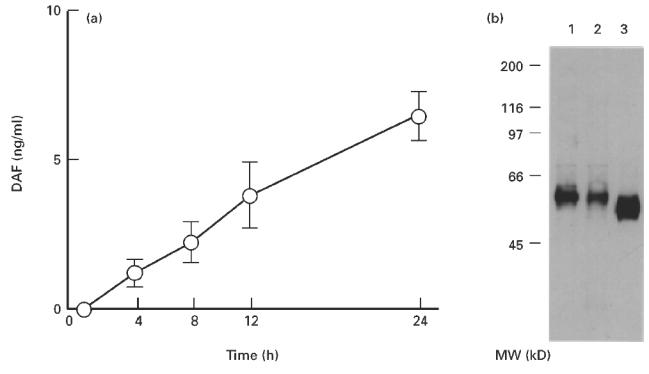
(a) Time-dependent increase in the amount of DAF released into the HT-29 culture supernatant. A supernatant of HT-29 monolayers cultured under standard culture conditions was collected at each time point, and the amount of DAF was determined by ELISA. Values shown are means ± s.e.m. of five experiments. (b) Immunoprecipitation of DAF protein in the culture supernatant of biotin surface-labelled HT-29 cells. The supernatant and cell lysate were immunoprecipitated with Sepharose beads coupled with 1C6 anti-DAF antibody. The Sepharose-bound biotinylated DAF was subjected to electrophoresis and blotted onto a nitrocellulose membrane. The membrane was then treated with horseradish peroxidase (HRP)-labelled streptavidin. Lane 1, membrane-associated DAF; lane 2, DAF spontaneously released into the culture supernatant; lane 3, DAF released with phosphatidylinositol-specific phospholipase C (PIPLC) treatment.
When the culture supernatant was centrifuged at 100 000 g, nearly a half of DAF was precipitated (Fig. 4), indicating that DAF in this fraction was present as a membrane-bound form and that the remainder in the supernatant was present as a soluble form. We then absorbed biotinylated proteins in the soluble form fraction with streptavidin-agarose beads. Sixteen percent of DAF was removed from this fraction with the streptavidin-beads (Table 1). Treatment of cells with cycloheximide did not change this ratio significantly. Because 73% of DAF was removed from the PIPLC-released material, in which DAF was supposed to be derived from the cell surface, the percentage of DAF derived from the cell surface in the soluble form fraction was estimated to be about 21%.
Fig. 4.
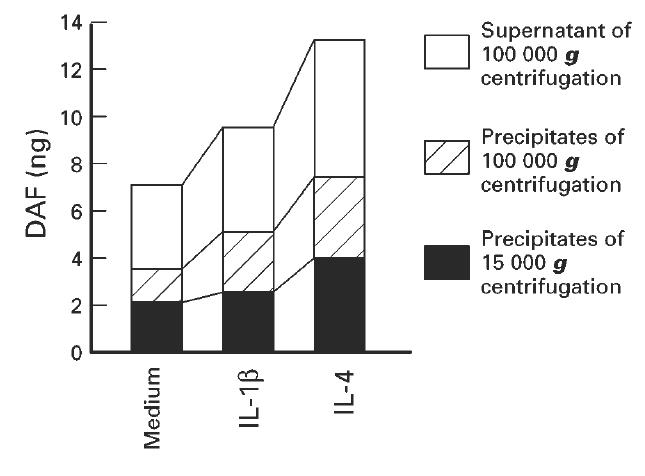
Recovery of DAF in the fractions separated by centrifugation of the culture supernatant of HT-29 cells. Cells were incubated for 24 h with or without addition of IL-1β (10 ng/ml) or IL-4 (100 ng/ml). The culture supernatant was centrifuged at 15 000 g for 15 min, and its supernatant was further centrifuged at 100 000 g for 60 min. DAF in each fraction was measured by use of ELISA. Values are expressed as the amount of DAF in 1 ml of the culture supernatant.
Table 1.
Streptavidin absorption of biotinylated DAF in the soluble form fraction of the culture supernatant and in the phosphatidylinositol-specific phospholipase C (PIPLC)-released material

IL-4 and IL-1β enhance release and expression of DAF on HT-29 cells
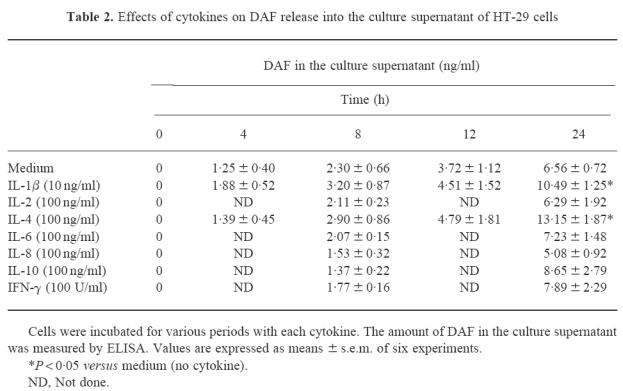
The effects of IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10 and IFN-γ on spontaneous DAF release from the cell surface of HT-29 cells were studied at various time points during incubation. Two of these, IL-1β and IL-4, induced an increase in the amount of DAF released into the culture supernatant (Table 2). After 24 h, IL-4 at a concentration of 100 ng/ml increased the level of DAF release 2.0-fold, and IL-1β at 10 ng/ml increased it 1.6-fold, and both increases were statistically significant (P < 0.05).
Table 2.
Effects of cytokines on DAF release into the culture supernatant of HT-29 cells
To examine the effects of these cytokines in detail, various concentrations were incubated with HT-29 cells for 24 h, and release and expression of DAF protein were determined (Fig. 5a). With IL-4 stimulation, an increase in DAF release was observed at 1 ng/ml and reached a maximum at 10 ng/ml. With IL-1β stimulation, a maximum increase was already observed at 1 ng/ml, but the effect was more pronounced with IL-4 than with IL-1β at concentrations of 100 ng/ml. The IL-4-dependent increase in DAF release was accompanied by an increase in PIPLC-sensitive cell surface DAF as well as by an increase in DAF in the remaining cell lysate (Fig. 5b,c). IL-1β-induced increases in these fractions were not so apparent as the IL-4-induced increase. In the culture supernatant, these cytokines increased DAF in both the precipitate and the supernatant fractions after 100 000 g centrifugation (Fig. 4). When we examined effects of IL-4 (100 ng/ml) and IL-1β (100 ng/ml) stimulation by flow cytometry, increase of the fluorescence intensity was not apparent (data not shown), probably because this method was not sensitive enough to detect the increase of cell surface DAF.
Fig. 5.
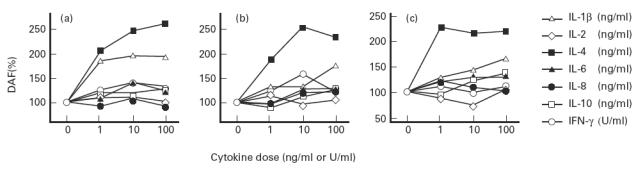
Effects of cytokines on the release and expression of DAF on HT-29 cells. Cells were incubated for 24 h with various concentrations of each cytokine. After each culture supernatant was collected, cells were detached and incubated with phosphatidylinositol-specific phospholipase C (PIPLC). After the medium was collected, the remaining cells were solubilized. The amounts of DAF in the culture supernatant, DAF released into the medium by PIPLC and DAF in the lysate of the remaining cells were determined by ELISA. (a) DAF which had shed into the culture supernatant. (b) PIPLC-sensitive cell surface DAF. (c) DAF in the lysate of the remaining cells. Values are expressed as a percentage of that of the control in which cytokines were not present. Values shown are means of four experiments.
Effects of actinomycin D, cycloheximide and brefeldin A on the IL-4- and IL-1β-stimulated increase of release and expression of DAF on HT-29 cells
We then investigated the effects of actinomycin D, cycloheximide and brefeldin A on the IL-4- and IL-1β-stimulated increase in release and expression of DAF. As shown in Fig. 6, treatment of cells with either actinomycin D 100 nm, cycloheximide 1 μm, or brefeldin A 0.5 ng/ml inhibited the increase in DAF release from the cell surface induced by IL-4 and IL-1β stimulation. Actinomycin D and cycloheximide also inhibited the IL-1β-induced increase in amounts of PIPLC-sensitive cell surface DAF and DAF in the cell lysate. However, the IL-4-induced increase was not inhibited by cycloheximide treatment at a concentration of 10 mm. Brefeldin A inhibited the IL-4- and IL-1β-induced increase in PIPLC-sensitive cell surface DAF, but not that of DAF in the cell lysate.
Fig. 6.
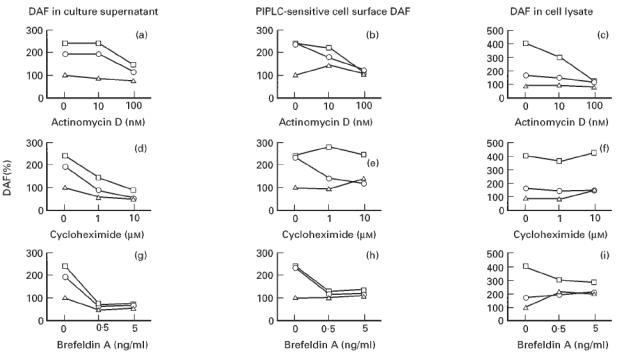
Effects of actinomycin D, cycloheximide, and brefeldin A on the IL-4- and IL-1β-stimulated increase in the release and expression of DAF. Cells were incubated for 24 h with IL-4 (10 ng/ml) or IL-1β (10 ng/ml) in the presence of various concentrations of actinomycin D (a–c), cycloheximide (d–f), or brefeldin A (g–i). The amounts of DAF shed into the culture supernatant (a,d,g), phosphatidylinositol-specific phospholipase C (PIPLC)-sensitive cell surface DAF (b,e,h), and DAF in the cell lysate (c,f,i) were then determined. Values are expressed as a percentage of that of the control in which the cytokines and inhibitors were absent. Values shown are means of three experiments. □, IL-4; ○, IL-1β; ▵, medium (without cytokine).
DISCUSSION
In this study, we demonstrated that cultured human intestinal epithelial cells, HT-29, constitutively express DAF on their cell surface and spontaneously release DAF into the culture supernatant, as described in various type of cells, such as HeLa cells [17,18], human polymorphonuclear cells [19] and human umbilical vein endothelial cells [20]. The results in this study also suggest that various mechanisms are involved in the release of DAF from HT-29 cells.
First, we found that nearly a half of DAF in the culture supernatant was present as a membrane-bound form. DAF of this form can be exported by non-specific solubilizations, such as the release of membrane fragments after cell death, or released in the form of intact vesicles budding from the cell surface, as some tumour cells are capable of vesiculating under certain conditions [21]. Biotinylated DAF in the culture supernatant with an apparent molecular weight similar to that of cell surface DAF was likely to represent DAF in this form.
The remaining half of DAF in the culture supernatant was present as a soluble form. When we absorbed DAF from the surface-biotinylated cells with streptavidin-beads, about one-fifth of DAF in the soluble form was likely to originate from the cell surface. This process should involve cleavage of DAF from the plasma membrane, such as by enzymatic hydrolysis of the GPI anchor. In the study of HeLa cells [18], a cell-associated GPI-specific phospholipase D was shown to be responsible for the cleavage and release of DAF from the cell surface. In this study, PIPLC treatment released DAF which was slightly smaller than DAF in the culture supernatant. A band of the PIPLC-released DAF appeared to correspond to the size of anchor-minus DAF as described in erythrocytes [22]. In HeLa cells, however, constitutively released DAF comigrated with PIPLC-released DAF [18]. We have no ready explanation for the discrepancy of the molecular weights of DAF cleaved by enzymatic hydrolysis. The cleavage of DAF from the cell membrane awaits further clarification.
The remaining four-fifths of DAF in the soluble form was not absorbed with streptavidin-beads, suggesting that this fraction of DAF was not released from the biotinylated cell surface. Because alternatively spliced forms of human DAF mRNA which may encode secreted forms of DAF have been reported [23], DAF without biotin label was possibly derived by secretion from within the cell.
Among the various cytokines examined in this study, we found that IL-4 and IL-1β enhanced the release of DAF from HT-29 cells. Actinomycin D, an inhibitor of RNA synthesis, inhibited the increase in DAF release as well as the expression of DAF protein. In contrast, when protein synthesis was inhibited by cycloheximide, the effects of IL-4 and IL-1β on DAF release were abrogated. However, there was no inhibition of the IL-4-enhanced expression of DAF protein. The reason for this discrepancy is unknown. Since IL-4 superinduces DAF mRNA in the presence of cycloheximide in HT-29 cells [8], one possible explanation is that cycloheximide at the concentration used in this study might not be sufficient to inhibit the translation of markedly increased ‘superinduced’ DAF mRNA. Brefeldin A, which blocks the intracellular transport of proteins, abrogated the effects of IL-4 and IL-1β on DAF release. Brefeldin A also inhibited the increase in DAF on the cell surface, but not in the cell lysate, suggesting that transport of the intracellular DAF protein to the cell surface is involved in this cytokine-induced increase in DAF release. In contrast to a strong increase of the amount of PIPLC-sensitive cell surface DAF as well as DAF in the lysate by IL-4, effects of IL-1β on DAF expression were modest, suggesting that IL-1β may exert its effect on DAF release by mainly increasing secretion of the protein.
We have recently shown that DAF is released into the colonic lumen of UC patients with active disease in relation to the severity of mucosal inflammation [5]. The finding suggests that the measurement of stool DAF is useful as a non-invasive means of monitoring intestinal disease activity in patients with UC. Although an increase in local IL-4 production has not been demonstrated [11,24,25], IL-1β mRNA expression is increased in actively inflamed UC tissues [26,27]. Our results suggest that these cytokines may play a role in the DAF release into the colonic lumen of UC patients.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Science, Sports and Culture, Tokyo, Japan.
References
- 1.Strober W, Ehrhardt RO. Chronic intestinal inflammation: an unexpected outcome in cytokine or T cell receptor mutant mice. Cell. 1993;75:203–5. doi: 10.1016/0092-8674(93)80062-j. [DOI] [PubMed] [Google Scholar]
- 2.Ueki T, Mizuno M, Uesu T, et al. Distribution of activated complement, C3b, and its degraded fragments, iC3b/C3dg, in the colonic mucosa of ulcerative colitis (UC) Clin Exp Immunol. 1996;104:286–92. doi: 10.1046/j.1365-2249.1996.17721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholson-Weller A, Burge J, Fearon DT, et al. Isolation of a human erythrocyte membrane glycoprotein with decay-accelerating activity for C3 convertases of the complement system. J Immunol. 1982;129:184–9. [PubMed] [Google Scholar]
- 4.Uesu T, Mizuno M, Inoue H, et al. Enhanced expression of decay accelerating factor and CD59/homologous restriction factor 20 on the colonic epithelium of ulcerative colitis. Lab Invest. 1995;72:587–91. [PubMed] [Google Scholar]
- 5.Inaba T, Mizuno M, Ohya S, et al. Decay-accelerating factor (DAF) in stool specimens as a marker of disease activity in patients with ulcerative colitis (UC) Clin Exp Immunol. 1998;112:237–41. doi: 10.1046/j.1365-2249.1998.00573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moutabarrik A, Nakanishi I, Namiki M, et al. Cytokine-mediated regulation of the surface expression of complement regulatory proteins, CD46 (MCP), CD55 (DAF), and CD59 on human vascular endothelial cells. Lymphokine Cytokine Res. 1993;12:167–72. [PubMed] [Google Scholar]
- 7.Moutabarrik A, Nakanishi I, Matsumoto M, et al. Human glomerular epithelial cells synthesize and secrete the third component of complement. Nephron. 1995;70:55–61. doi: 10.1159/000188544. [DOI] [PubMed] [Google Scholar]
- 8.Andoh A, Fujiyama Y, Sumiyoshi K, et al. Interleukin 4 acts as an inducer of decay-accelerating factor gene expression in human intestinal epithelial cells. Gastroenterology. 1996;111:911–8. doi: 10.1016/s0016-5085(96)70058-6. [DOI] [PubMed] [Google Scholar]
- 9.Bjørge L, Jensen TS, Matre R. Characterisation of the complement-regulatory proteins decay-accelerating factor (DAF, CD55) and membrane cofactor protein (MCP, CD46) on a human colonic adenocarcinoma cell line. Cancer Immunol Immunother. 1996;42:185–92. doi: 10.1007/s002620050269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sartor RB. Cytokines in intestinal inflammation: pathophysiological and clinical considerations. Gastroenterology. 1994;106:533–9. doi: 10.1016/0016-5085(94)90614-9. [DOI] [PubMed] [Google Scholar]
- 11.Niessner M, Volk BA. Altered Th1/Th2 cytokine profiles in the intestinal mucosa of patients with inflammatory bowel disease as assessed by quantitative reversed transcribed polymerase chain reaction (RT-PCR) Clin Exp Immunol. 1995;101:428–35. doi: 10.1111/j.1365-2249.1995.tb03130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujita T, Inoue T, Ogawa K, et al. The mechanism of action of decay-accelerating factor (DAF). DAF inhibits the assembly of C3 convertases by dissociating C2a and Bb. J Exp Med. 1987;166:1221–8. doi: 10.1084/jem.166.5.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizuno M, Nakagawa M, Uesu T, et al. Detection of decay-accelerating factor in stool specimens of patients with colorectal cancer. Gastroenterology. 1995;109:826–31. doi: 10.1016/0016-5085(95)90390-9. [DOI] [PubMed] [Google Scholar]
- 14.Medof ME, Walter EI, Roberts WL, et al. Decay accelerating factor of complement is anchored to cells by a C-terminal glycolipid. Biochemistry. 1986;25:6740–7. doi: 10.1021/bi00370a003. [DOI] [PubMed] [Google Scholar]
- 15.Lisanti MP, Sargiacomo M, Graeve L, et al. Polarized apical distribution of glycosyl-phosphatidylinositol-anchored proteins in a renal epithelial cell line. Proc Natl Acad Sci USA. 1989;85:9557–61. doi: 10.1073/pnas.85.24.9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 17.Medof ME, Walter EI, Rutgers JL, et al. Identification of the complement decay-accelerating factor (DAF) on epithelium and glandular cells and in body fluids. J Exp Med. 1987;165:848–64. doi: 10.1084/jem.165.3.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metz CN, Brunner G, Choi Muira NH, et al. Release of GPI-anchored membrane proteins by a cell-associated GPI-specific phospholipase D. EMBO J. 1994;13:1741–51. doi: 10.1002/j.1460-2075.1994.tb06438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tausk F, Fey M, Gigli I. Endocytosis and shedding of the decay accelerating factor on human polymorphonuclear cells. J Immunol. 1989;143:3295–302. [PubMed] [Google Scholar]
- 20.Tsuji S, Kaji K, Nagasawa S. Decay-accelerating factor on human umbilical vein endothelial cells. Its histamine-induced expression and spontaneous rapid shedding from the cell surface. J Immunol. 1994;152:1404–10. [PubMed] [Google Scholar]
- 21.Masella R, Cantafora A, Guidoni L, et al. Characterization of vesicles, containing an acylated oligopeptide, released by human colon adenocarcinoma cells. NMR and biochemical studies. FEBS Letters. 1989;246:25–29. doi: 10.1016/0014-5793(89)80246-7. [DOI] [PubMed] [Google Scholar]
- 22.Davitz MA, Low MG, Nussenzweig V. Release of decay-accelerating factor (DAF) from the cell membrane by phosphatidylinositol-specific phospholipase C (PIPLC). Selective modification of a complement regulatory protein. J Exp Med. 1986;163:1150–61. doi: 10.1084/jem.163.5.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caras IW, Davitz MA, Rhee L, et al. Cloning of decay-accelerating factor suggests novel use of splicing to generate two proteins. Nature. 1987;325:545–9. doi: 10.1038/325545a0. [DOI] [PubMed] [Google Scholar]
- 24.Karttunnen R, Breese EJ, Walker Smith JA, et al. Decreased mucosal interleukin-4 (IL-4) production in gut inflammation. J Clin Pathol. 1994;47:1015–8. doi: 10.1136/jcp.47.11.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen OH, Koppen T, Rudiger N, et al. Involvement of interleukin-4 and -10 in inflammatory bowel disease. Dig Dis Sci. 1996;41:1786–93. doi: 10.1007/BF02088746. [DOI] [PubMed] [Google Scholar]
- 26.Isaacs KL, Sartor RB, Haskill S. Cytokine messenger RNA profiles in inflammatory bowel disease mucosa detected by polymerase chain reaction amplification. Gastroenterology. 1992;103:1587–95. doi: 10.1016/0016-5085(92)91182-4. [DOI] [PubMed] [Google Scholar]
- 27.McCabe RP, Secrist H, Botney M, et al. Cytokine mRNA expression in intestine from normal and inflammatory bowel disease patients. Clin Immunol Immunopathol. 1993;66:52–8. doi: 10.1006/clin.1993.1007. [DOI] [PubMed] [Google Scholar]