

Abstract
In susceptible H-2s mice, mercuric chloride (HgCl2) induces an autoimmune syndrome characterized by production of anti-nucleolar antibodies (ANoA) and increased serum levels of IgG1 and IgE antibodies. The increase in serum IgG1 and IgE, which are under IL-4 control, suggests a role for the Th2 subset in the induction of this syndrome. We have previously shown that administration of IL-12, a potent Th1-promoting cytokine, resulted in a dramatic reduction of the HgCl2-induced anti-nucleolar antibody titres and inhibited serum IgG1 increase. These results suggest that Th1 T cells can down-regulate ANoA, and support a role for the Th2 subset in ANoA production, possibly via IL-4. To examine the role of IL-4 in this syndrome, C57Bl/6 mice (H-2b) with a targeted deletion of the IL-4 gene were mated with A.SW mice (H-2s) to yield H-2s mice lacking IL-4. We then analysed ANoA and serum immunoglobulin levels in these mice after HgCl2 treatment. While mercury-treated IL-4−/− H-2s mice had virtually no detectable serum IgG1 or IgE, and very low levels of IgG1 ANoA, these mice had levels of IgG2a and IgG2b class ANoA comparable to mercury-treated IL-4+ H-2s mice, indicating that IL-4 is not required for the ANoA response in mercury-induced autoimmunity.
Keywords: mercury, autoimmunity, IL-4, anti-nucleolar antibodies
INTRODUCTION
Mouse or rat strains expressing certain MHC antigens are exquisitely susceptible to the heavy metal induction of a complex autoimmune syndrome [1–4]. In H-2s mice, subtoxic doses of mercuric chloride (HgCl2) induce an autoimmune dysfunction characterized by the production of anti-nucleolar autoantibodies (ANoA) and hyperglobulinaemia (especially pronounced for IgG1 and IgE). These increases in serum immunoglobulin peak 2–3 weeks after the beginning of the Hg injections, whereas ANoA can persist for several months after the induction phase. In mice, susceptibility to mercury is dependent upon the expression of class II I-As molecules [5–7].
The T cell regulation of this syndrome is not fully understood. The increase in serum IgG1 and IgE in Hg-treated mice suggests that Th2 cells play an important role in the pathogenesis of this syndrome [3,8]. In susceptible strains, HgCl2 can directly induce secretion of IL-4, an important Th2 cytokine which promotes isotype switching to IgG1 and IgE [9–13]. However, interventions favouring a Th1-type response yield differing results. We have previously shown in A.SW mice (H-2s) that administration of IL-12 before treatment with HgCl2 results in reduced serum ANoA levels and a partially reduced IgG1 response, but has no effect on the serum IgE increase [14]. Conversely, Doth et al. have shown that pretreatment of HgCl2-injected B10.S mice (H-2s) with interferon-gamma (IFN-γ) leads to decreased serum IgE levels, but has no effect on serum ANoA [15]. While treatment of A.SW mice with anti-IL-4 MoAb inhibits the mercury induction of serum IgG1 and IgE levels, it increases ANoA isotype of subclasses other than IgG1 [16]. Thus, while Th2 cells seem to be associated with induction and Th1 cells with resolution, the specific contributions of these subsets to mercury-induced autoimmunity remain unclear.
To define clearly the role of IL-4 in this syndrome, C57Bl/6 mice (H-2b) with a targeted deletion of the IL-4 gene were mated with A.SW mice (H-2s) to yield H-2s mice lacking IL-4. These mice were then treated with mercury and serum ANoA, IgE and IgG levels were analysed. The present study indicates that, in mercury-induced autoimmunity, IL-4 is required for the serum increases in IgG1 and IgE, but not for ANoA production.
MATERIALS AND METHODS
Breeding of IL-4−/− I-As mice
Female A.SW/SnJ (H-2s) mice and male C57Bl/6 IL-4−/− (H-2b) mice were obtained from the Jackson Laboratories (Bar Harbor, ME) and maintained in our animal facilities. C57Bl/6 IL-4−/− males were mated with A.SW females and the resulting F1 progeny, which was IL-4+/– and H-2b/s, was interbred in brother–sister matings. F2 mice were then ear-punched for identification and polymerase chain reaction (PCR) genotyping was performed on ear biopsies. To identify I-Ab, I-As, and I-Ab/s mice, we devised two pairs of primers that amplified 213-bp sequences corresponding to the first domain of either the I-Ab or I-Asα-chain. Each primer differed by two nucleotides at the 3′ end from its counterpart designed to amplify the other allele and, in pilot experiments, each set of primers specifically amplified the appropriate sequence, but not its opposite allele (not shown). Primer sequences were as follows: I-Ab sense, TGA GGC CGA CCA CGT AGG CAC; I-Ab antisense, TCC CAA GTT GTG TTT TAC TAC; I-As sense, TGA GGC CGA CCA CGT AGG CGT; I-As antisense, TCC CAA GGT GTA TTT TCC TGT. To identify targeted and wild-type IL-4 genes, we used the PCR primer sequences and conditions that were recommended by Jackson Labs (http://lena.jax.org/resources/documents/imr/protocols/Il4_KO.html). Irrespective of their IL-4 genotype, only mice that were demonstrated to be I-As homozygous by PCR were fully evaluated in subsequent experiments.
HgCl2 treatment
Groups of at least 2-month-old mice were injected three times a week subcutaneously with 30 μg HgCl2 (Sigma Chemical Co., St Louis, MO) in 100 μl sterile PBS throughout the duration of the experiment [16]. Blood was obtained weekly by retro-orbital bleeding.
ANoA immunofluorescence
ANoA levels in serially diluted mouse serum were determined by indirect immunofluorescence as previously described [14,17].
ELISA for mouse serum IgG1, IgG2a, IgG2b and IgE
Total serum IgG1, IgG2a, IgG2b and IgE levels were determined using a sandwich ELISA as previously described [14].
RESULTS
ANoA production in mercury-treated IL-4+ and IL-4−/− I-As mice
Groups of IL-4+/+, IL-4+/– or IL-4−/− mice were injected with HgCl2 as described in Materials and Methods and their sera were tested for nucleolar reactivity by immunofluorescence (IF). The I-As animals evaluated in this study developed IgG ANoA, whereas no ANoA were detected in sera from C57Bl/6 IL-4−/− or IL-4+/+ mice at any time point (data not shown), confirming the requirement of the H-2s haplotype for susceptibility to Hg-induced autoimmunity. IgG1 ANoA were detected in the sera of IL-4+/+ and IL-4+/– mice after week 2 and eventually reached titres ≥ 10 000 (Fig. 1). In these and all subsequent experiments, there was no statistically significant difference in responses between IL-4+/+ mice and IL-4+/– mice, therefore these animals were pooled into a single group (referred to hereafter as IL-4+). In contrast to the IL-4+ group, IgG1 ANoA were not detected in the IL-4−/− group until week 3. ANoA levels in this group then slowly increased, but their mean titre remained very low (≤ 300). IgG2a and IgG2b ANoA were present after 2 weeks in sera from IL-4+ mice, with peak titres attained at weeks 4 (8750) and 6 (2214) for IgG2a and IgG2b, respectively. IL-4−/− mice also developed IgG2a and IgG2b ANoA at levels comparable to those of the IL-4+ group (Fig. 1).
Fig. 1.

Anti-nucleolar antibodies (ANoA) in mercury-treated IL-4+ (▪) and IL-4−/− mice (□). ANoA were detected by immunofluorescence on HEp-2 cells using isotype-specific FITC conjugates [14]. Results are expressed as serum titres (inverse of the highest serum dilution that yielded nucleolar fluorescence). Eight mice were included in the IL-4+ group and four mice in the IL-4−/− group. Comparisons between the two groups were performed using the Mann–Whitney test. *Statistically significant (P < 0.05) differences.
Serum IgG and IgE isotype levels in IL-4+ and IL-4−/− I-As mice
To determine the total serum levels for IgG1, IgG2a and IgG2b in Hg-treated IL-4+ and IL-4−/− mice, serum dilutions were tested using a sandwich ELISA. Levels of IgG1 in IL-4+ mice increased approx. three-fold after week 2 of HgCl2 treatment (Fig. 2). Serum levels peaked at week 3 and declined rapidly afterwards. However, no overall change in IgG1 levels was seen in the IL-4−/− group, with levels remaining similar to those of week 0. Basal levels of IgG1 in IL-4−/− mice were also significantly lower than those of IL-4+ mice: 0.1540 mg/ml versus 0.3129 mg/ml (P = 0.004 using the Mann–Whitney test). No differences were seen in IgG2a and IgG2b levels between the two groups throughout the course of the experiment. Similarly, total serum IgE was measured in sera by sandwich ELISA. While IL-4+ mice showed a dramatic increase in serum IgE by week 2 (approx. 23-fold increase) compared with pre-injection levels, virtually no IgE was detected in sera from IL-4−/− mice throughout the course of the experiment.
Fig. 2.
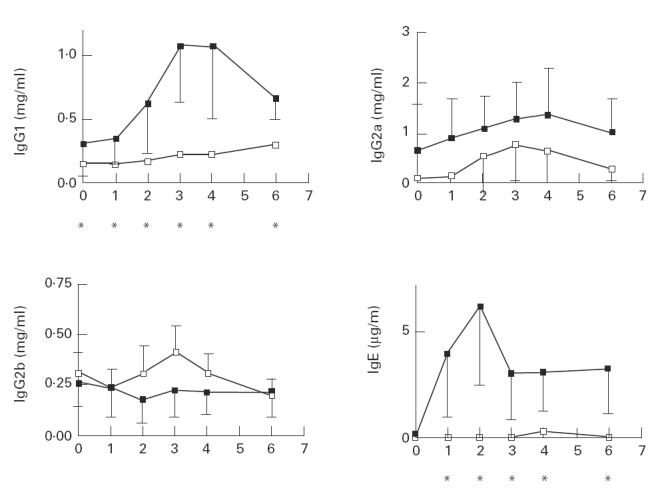
Serum immunoglobulin levels in mercury-treated IL-4+ (▪) and IL-4−/− mice (□). Quantification of immunoglobulin isotypes was performed using a sandwich ELISA as described previously [14]. Eight mice were included in the IL-4+ group and four mice in the IL-4−/− group. Comparisons between the two groups were performed using the Mann–Whitney test. *Statistically significant (P < 0.05) differences.
DISCUSSION
The study of murine Hg-induced autoimmunity is important because of the many reported associations between various drugs, chemicals, or pollutants and human autoimmune diseases [18,19]. Heavy metals are frequent environmental pollutants and Hg is also contained in various creams and ointments. Exposure to Hg can result in hypersensitivity reactions and in membranous glomerulonephritis with IgG deposits [20]. The levels of mercury vapour required to induce autoimmunity in H-2s mice are similar to those found in occupationally exposed humans [21]. Workers exposed to mercury have increased levels of serum IgE or antinuclear autoantibodies even with urinary Hg levels within admitted ‘safe’ limits [22,23].
Anti-nucleolar antibodies are not only induced by mercury in H-2s mice, but in humans they are found almost exclusively in scleroderma, the systemic autoimmune disease most frequently associated with environmental factors [24]. Indeed, exposure to chemicals or pollutants (silica, epoxy resins, biogenic amines, urea formaldehyde, and various organic chemicals) has been implicated in the aetiology of scleroderma [25,26]. As in murine Hg-induced autoimmunity, certain MHC haplotypes are associated with the development of chemically induced autoimmunity in humans. For instance, DR3 predisposes to glomerulonephritis in patients treated with gold salts. DR5 is a susceptibility factor for the development of scleroderma in chemically exposed individuals [18,19].
The present study shows that, while mercury-treated H-2s IL-4−/− mice had undetectable serum levels of IgE and very low levels of IgG1 class ANoA, levels of IgG2a and IgG2b class ANoA remained similar to those of IL-4+ mice. This confirms that IL-4 is required for the isotype switching to IgG1 and IgE in mercury-induced autoimmunity, but not required for the loss of tolerance to nucleolar antigens. In contrast, no differences in total serum levels of IgG2a and IgG2b class antibodies were seen in IL-4−/− mice compared with IL-4+ mice.
Several authors have suggested that susceptibility of rodents to develop systemic autoimmunity in response to metals is associated with the development of Th2 responses [8,27], and resistance to disease is associated with a Th1 response [28]. Previous findings in our laboratory demonstrating that administration of IL-12 prior to HgCl2 injection resulted in down-regulation of ANoA levels would seem to support this hypothesis. However, Hultman et al. recently reported that mice of the strain S.JL (H-2s), which has a pronounced deficiency in Th2-promoting CD4+ NK1.1+ T cells, lacked the increase in IL-4+ cells, but developed a systemic autoimmunity very similar to that in A.SW mice after treatment with HgCl2 [29]. The present observations are in agreement with these findings and indicate that a predominantly Th2 response is not required for induction of autoimmunity by HgCl2.
While Ochel et al. previously demonstrated that administration of HgCl2 to anti-IL-4 antibody-treated H-2s mice resulted in a shift in the ANoA isotype profile to that of IgG2a, IgG2b and IgG3 [16], we did not see a compensatory increase in the other ANoA subclasses in IL-4−/− mice. One possible explanation for these differing results is that use of neutralizing anti-IL-4 antibodies resulted only in an incomplete or short-term inactivation of IL-4, thereby yielding differing results. The block in isotype switching to IgG1 did not result in an accumulation of IgM autoantibody-producing B cells, since we did not detect IgM ANoA in any animal group (data not shown). Overall, our observations parallel those made in a conventional immune response by Kuhn et al. in IL-4−/− mice, where immunization with nitrophenyl-chicken γ-globulin results in low levels of IgG1 and IgE, while the IgG2a, IgG2b and IgG3 responses are preserved [30].
Several lines of evidence suggest independent regulation of the different manifestations of the disease. For example, while administration of IL-4 MoAb [16] or targeted deletion of the IL-4 gene resulted in decreases in serum IgG1 and IgE in mercury-treated mice, neither approach abrogated the loss of tolerance to nucleolar antigens and the resultant ANoA production. Similarly, administration of IL-12 to mercury-treated mice, while down-regulating ANoA production and partially preventing IgG1 increase, did not inhibit IgE induction in this model [14]. Thus, it is unlikely that a single regulatory mechanism exists which controls all the various aspects of the syndrome.
While Th1/Th2 cytokines play an important role in the B cell activation and isotype switching seen in Hg-induced autoimmunity, it is likely that the loss of tolerance to nucleolar antigens arises by a mechanism independent of conventional Th1/Th2 regulation. Serum increases in IgG1, IgE and IgG2a probably reflect the activation of various T cell subsets in this model, while the antigen-specific phase of the disease (ANoA production) may be regulated by factors such as antigen modification and MHC antigen presentation pathways [31]. Thus, interventions such as rIFN-γ or anti-IL-4 MoAb treatments affect isotype switching and total serum immunoglobulin levels, but do not prevent loss of tolerance.
Note added in proof
Since the submission of this manuscript, similar findings have been reported in IL-4-deficient mice by Kono et al. (J Immunol 1998; 161:234–40).
Acknowledgments
This study was supported by NIH grant R01 AI-26665.
REFERENCE
- 1.Druet P, Sheela R, Pelletier L. Th1 and Th2 cells in autoimmunity. Chem Immunol. 1996;63:138–70. [PubMed] [Google Scholar]
- 2.Mathieson PW. Mercuric chloride-induced autoimmunity. Autoimmunity. 1992;13:243–7. doi: 10.3109/08916939209004830. [DOI] [PubMed] [Google Scholar]
- 3.Griem P, Gleichmann E. Metal ion induced autoimmunity. Curr Opin Immunol. 1995;7:831–8. doi: 10.1016/0952-7915(95)80056-5. [DOI] [PubMed] [Google Scholar]
- 4.Hanley GA, Schiffenbauer J, Sobel ES. Class II haplotype differentially regulates immune response in HgCl2-treated mice. Clin Immunol Immunopathol. 1997;84:328–37. doi: 10.1006/clin.1997.4405. [DOI] [PubMed] [Google Scholar]
- 5.Mirtcheva J, Pfeiffer C, De Bruijn JA, Jacquesmart F, Gleichmann E. Immunological alterations induced by mercury compounds. III. H-2A acts as an immune response and H-2E as an immune response ‘suppression’ locus for HgCl2-induced antinucleolar antibodies. Eur J Immunol. 1989;19:2257–61. doi: 10.1002/eji.1830191212. [DOI] [PubMed] [Google Scholar]
- 6.Hultman P, Bell LJ, Enestrom S, Pollard KM. Murine susceptibility to mercury. I. Autoantibody profiles and systemic immune deposits in inbred, congenic, and intra-H-2 recombinant strains. Clin Immunol Immunopathol. 1992;65:98–109. doi: 10.1016/0090-1229(92)90212-7. [DOI] [PubMed] [Google Scholar]
- 7.Hultman P, Bell LJ, Enestrom S, Pollard KM. Murine susceptibility to mercury. II. Autoantibody profiles and renal immune deposits in hybrid, backcross, and H-2d congenic mice. Clin Immunol Immunopathol. 1993;68:9–20. doi: 10.1006/clin.1993.1088. [DOI] [PubMed] [Google Scholar]
- 8.Goldman M, Druet P, Gleichmann E. TH2 cells in systemic autoimmunity: insights from allogeneic diseases and chemically-induced autoimmunity. Immunol Today. 1991;12:223–7. doi: 10.1016/0167-5699(91)90034-Q. [DOI] [PubMed] [Google Scholar]
- 9.Prigent P, Saoudi A, Pannetier C, et al. Mercuric chloride, a chemical responsible for T helper cell (Th)2-mediated autoimmunity in Brown Norway rats, directly triggers T cells to produce interleukin-4. J. Clin Invest. 1995;96:1484–9. doi: 10.1172/JCI118185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillespie KM, Qasim FJ, Tibbatts LM, Thiru S, Oliveira DB, Mathieson PW. Interleukin-4 gene expression in mercury-induced autoimmunity. Scand J Immunol. 1995;41:268–72. doi: 10.1111/j.1365-3083.1995.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 11.Gillespie KM, Saoudi A, Kuhn J, et al. Th1/Th2 cytokine gene expression after mercuric chloride in susceptible and resistant rat strains. Eur J Immunol. 1996;26:2388–92. doi: 10.1002/eji.1830261018. [DOI] [PubMed] [Google Scholar]
- 12.Badou A, Savignac M, Moreau M, et al. HgCl2-induced interleukin-4 gene expression in T cells involves a protein kinase C-dependent calcium influx through L-type calcium channels. J Biol Chem. 1997;272:32411–8. doi: 10.1074/jbc.272.51.32411. [DOI] [PubMed] [Google Scholar]
- 13.Oliveira DB, Gillespie K, Wolfreys K, Mathieson PW, Qasim F, Coleman JW. Compounds that induce autoimmunity in the Brown Norway rat sensitize mast cells for mediator release and interleukin-4 expression. Eur J Immunol. 1995;25:2259–64. doi: 10.1002/eji.1830250822. [DOI] [PubMed] [Google Scholar]
- 14.Bagenstose LM, Salgame P, Monestier M. IL-12 down-regulates autoantibody production in mercury-induced autoimmunity. J Immunol. 1998;160:1612–7. [PubMed] [Google Scholar]
- 15.Doth M, Fricke M, Nicoletti F, et al. Genetic differences in immune reactivity to mercuric chloride (HgCl2): immunosuppression of H-2d mice is mediated by interferon-gamma (IFN-gamma) Clin Exp Immunol. 1997;109:149–56. doi: 10.1046/j.1365-2249.1997.4041300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochel M, Vohr HW, Pfeiffer C, Gleichmann E. IL-4 is required for the IgE and IgG1 increase and IgG1 autoantibody formation in mice treated with mercuric chloride. J Immunol. 1991;146:3006–11. [PubMed] [Google Scholar]
- 17.Monestier M, Losman MJ, Novick KE, Aris JP. Molecular analysis of mercury-induced antinucleolar antibodies in H-2s mice. J Immunol. 1994;152:667–75. [PubMed] [Google Scholar]
- 18.Bigazzi P. Autoimmunity induced by chemicals. Clin Toxicol. 1988;26:125–56. doi: 10.3109/15563658809000343. [DOI] [PubMed] [Google Scholar]
- 19.Gleichmann E, Kimber I, Purchase IFH. Immunotoxicology: suppressive and stimulatory effects of drugs and environmental chemicals on the immune system. Arch Toxicol. 1989;63:257–73. doi: 10.1007/BF00278639. [DOI] [PubMed] [Google Scholar]
- 20.Goyer RA. Environmentally related diseases of the urinary tract. Med Clin North Am. 1990;74:377–89. doi: 10.1016/s0025-7125(16)30568-5. [DOI] [PubMed] [Google Scholar]
- 21.Warfvinge K, Hansson H, Hultman P. Systemic autoimmunity due to mercury vapor exposure in genetically susceptible mice: dose–response studies. Toxicol Appl Pharmacol. 1995;132:299–309. doi: 10.1006/taap.1995.1111. [DOI] [PubMed] [Google Scholar]
- 22.Cardenas A, Roels H, Bernard AM, et al. Markers of early renal changes induced by industrial pollutants. I. Application to workers exposed to mercury vapors. Br J Ind Med. 1998;50:17–27. doi: 10.1136/oem.50.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dantas DC, Queiroz ML. Immunoglobulin E and autoantibodies in mercury-exposed workers. Immunopharmacol Immunotoxicol. 1997;19:383–92. doi: 10.3109/08923979709046983. [DOI] [PubMed] [Google Scholar]
- 24.Monestier M. Nucleolar autoantibodies. In: Peter JB, Shoenfeld Y, editors. Autoantibodies. Amsterdam: Elsevier; 1996. pp. 567–73. [Google Scholar]
- 25.Rothfield NF. Autoantibodies in scleroderma. Rheum Dis Clin North Am. 1992;18:483–98. [PubMed] [Google Scholar]
- 26.Silman AJ. Epidemiology of scleroderma. Ann Rheum Dis. 1991;50:846–53. doi: 10.1136/ard.50.suppl_4.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathieson PW, Thiru S, Oliveira DB. Regulatory role of OX22high T cells in mercury-induced autoimmunity in the brown Norway rat. J Exp Med. 1993;177:1309–16. doi: 10.1084/jem.177.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castedo M, Pelletier L, Pasquier R, Druet P. Improvement of TH1 functions during the regulation phase of mercury disease in brown Norway rats. Scand J Immunol. 1994;39:144–50. doi: 10.1111/j.1365-3083.1994.tb03353.x. [DOI] [PubMed] [Google Scholar]
- 29.Johansson U, Sander B, Hultman P. Effects of the murine genotype on T cell activation and cytokine production in murine mercury-induced autoimmunity. J Autoimmun. 1997;10:347–55. doi: 10.1006/jaut.1997.0149. [DOI] [PubMed] [Google Scholar]
- 30.Kuhn R, Rajewsky K, Muller W. Generation and analysis of interleukin-4 deficient mice. Science. 1991;254:707–10. doi: 10.1126/science.1948049. [DOI] [PubMed] [Google Scholar]
- 31.Kubicka-Muranyi M, Griem P, Lubben B, Rottmann N, Luhrmann R, Gleichmann E. Mercuric-chloride-induced autoimmunity in mice involves up-regulated presentation by spleen cells of altered and unaltered nucleolar self antigen. Int Arch Allergy Immunol. 1995;108:1–10. doi: 10.1159/000237110. [DOI] [PubMed] [Google Scholar]