

Abstract
Anaphylaxis denotes an immediate hypersensitivity reaction to allergen, exclusively mediated by IgE antibodies. However, IgE antibodies do not explain all the syndromes that are encountered. We investigated potent IgG-mediated anaphylaxis in CD40-deficient mice that lack the immunoglobulin class switching for T cell-dependent antigens. Immunization with ovalbumin did not induce either humoral responses of IgG, IgA, and IgE, or systemic anaphylaxis in CD40-deficient mice. Although systemic anaphylaxis by active immunization was not observed in CD40-deficient mice, both passive cutaneous anaphylaxis (PCA) and passive systemic anaphylaxis assessed by mouse blood pressure monitoring with cervical artery catheterization did take place when antigen-specific IgG was transferred and then antigen challenge given. Further, to investigate the inflammatory pathway of IgG-mediated immediate hypersensitivity reactions, we focused on the Fcγ receptor (FcγR) function. Pretreatment of the mice with the anti-FcγRII/FcγRIII MoAb clearly blocked the response of PCA and passive systemic anaphylaxis, suggesting that they were initiated through FcγR. In conclusion, we directly demonstrate the IgG-mediated anaphylaxis and its triggering mechanism through FcγR in in vivo conditions. In addition to IgE-mediated anaphylaxis, IgG-mediated anaphylaxis should be considered and the blocking of FcγR would provide one of the therapeutic targets for the control of IgG-mediated hypersensitivity diseases.
Keywords: anaphylaxis, IgG, Fcγ receptor, type I hypersensitivity reaction, CD40
INTRODUCTION
Although signalling through high-affinity receptor for IgE (FcɛRI) initiated by cross-linking of the receptors through antigen–IgE antibody interaction is the major cause of anaphylaxis [1], IgE-independent mechanisms have been speculated to induce the syndrome [2–5]. Oettgen et al. reported that non-IgE stimulus played a role in anaphylaxis in in vivo condition from the results of active anaphylaxis in IgE-deficient mice [5]. However, it has not been directly determined what played a major role in IgE-independent anaphylaxis in vivo. In this study we attempt to determine the immunoglobulin class which is responsible for IgE-independent anaphylaxis in vivo. As it is difficult to obtain mice which lack the humoral responses of both IgE and IgG, or IgE and IgA, we used CD40-deficient mice which lack the immunoglobulin class switching and germinal centre formation for T cell-dependent antigens, resulting in lack of humoral responses of IgG, IgE, and IgA, but not IgM [6]. To investigate the role of immunoglobulin class in anaphylaxis, we induced systemic anaphylaxis in CD40-deficient mice.
As defined by Coombs & Gell, hypersensitivity reactions have been subdivided into four types [7]. Type I reactions, including anaphylaxis, are defined as immediate hypersensitivity mediated by mast cell-bound IgE antibody. Type II reactions are antibody- or complement-dependent cytotoxicities mediated by interaction of cell-bound antigen with IgG or IgM antibody. Type III reactions are known as immune complex tissue injury mediated by antigen–IgG immune complex and activated complement. However, recent reports have demonstrated the important role of Fcγ receptor (FcγR) in initiating IgG-mediated inflammatory reactions both in type II and in type III reactions and it may lead to a redefinition of inflammatory pathways [8–10]. In this study, we also investigate the inflammatory pathway of IgE-independent systemic anaphylaxis or the potent IgG-mediated anaphylaxis, and the role of FcγR function in type I hypersensitivity reaction in in vivo conditions.
MATERIALS AND METHODS
Mice
C57Bl/6 mice (H-2b) were supplied by the Institute for Laboratory Animal Research of Nagoya University School of Medicine. CD40-deficient mice were generated by gene targetting technique as previously reported [6]. A CD40+/− mouse was produced by backcrossing the originally described CD40−/− mouse to a C57Bl/6 mouse. The heterozygous litter mates were intercrossed to generate CD40+/+, CD40+/− and CD40−/− mice. These mice were genotyped by polymerase chain reaction (PCR) of genomic DNA obtained from tail biopsy using primers to identify the rearranged CD40 locus [6]. CD40+/+ wild-type mice were used as control.
Immunization and ELISA for antigen-specific immunoglobulins
Wild-type and CD40-deficient mice were immunized with 0.1 mg ovalbumin (OVA) and 1 mg aluminium hydroxide intraperitoneally on day 1, and boosted on day 15. Aluminium hydroxide (1 mg) alone was administered to wild-type mice as control. For the detection of OVA-specific antibodies, serum samples were added to 96-well flat-bottomed microtitre plates coated with OVA, and bound antibodies were detected by alkaline phosphatase-labelled isotype-specific antibodies for murine IgM, IgG1, IgG2a, IgG2b, IgG3 (Southern Biotechnology Associates, Birmingham, AL) and IgA (PharMingen, San Diego, CA). After addition of p-nitrophenyl phosphate as substrate, the optical density (OD) of each well was measured with a microplate spectrophotometer (EAR 400AT; SLT-Labinstruments, Salzburg, Austria) equipped with a 405-nm filter. As for the detection of OVA-specific IgE antibody, biotinylated anti-IgE MoAb (gift of Y. Hattori, Kowa Research Institute, Kowa Co., Tsukuba, Japan) was used. After addition of avidin–horseradish peroxidase and o-phenylenediamine dihydrochloride, the OD of each well was measured with the microplate spectrophotometer at 492 nm. The spectrophotometer was calibrated at 0 absorption using wells that were treated with the same as experimental wells except for the addition of mouse serum.
Purification of IgG fraction
After wild-type mice were immunized with OVA, anti-OVA serum was collected by repeated tail vessel cutting. IgG was purified by affinity chromatography using protein G column (HiTrap Protein G; Pharmacia, Uppsala, Sweden). After applying the serum onto the column and washing with washing buffer (20 mm Na-phosphate, pH 7.0), bound IgG was eluted to 1-ml fractions with elution buffer (0.1 m glycine–HCl, pH 2.7). Eluted IgG fractions were evaluated in amount by OD at 280 nm, and the fractions around the maximum peak were pooled and used in the following procedure. The buffer was exchanged for PBS using gel filtration method (NAPTM-25 Column; Pharmacia) and IgG was concentrated by ultrafiltration device (Centriprep-30; Amicon, Beverly, MA). Protein was quantified by a dye-binding protein assay (BioRad Protein Assay; BioRad, Richmond, CA) and diluted to 8 mg protein/ml by the appropriate PBS. Finally, the product was incubated at 56°C for 2 h to deplete binding capacity of IgE to FcɛRI, even if contaminating IgE was present. Purity of the anti-OVA IgG was then assessed by ELISA and no detectable IgE or IgA was confirmed.
Passive cutaneous anaphylaxis
Wild-type and CD40-deficient mice were injected intradermally on their backs with the purified mouse anti-OVA IgG fraction ranging from 0.8 μg to 0.025 μg per site in steps of two-fold dilution. One hour later the mice were injected intravenously with 0.25 mg OVA and 0.5% Evans blue, and their passive cutaneous anaphylaxis (PCA) titres were determined by extravasation of blue dye 30 min after antigen challenge [11,12]. The minimum dose which caused a blue spot at the injected site > 5 mm in diameter was defined as the PCA titre.
Assessment of systemic anaphylaxis
For assessment of active systemic anaphylaxis, mice were given 0.5 mg OVA intravenously to induce systemic anaphylaxis under light ether anaesthesia on day 29 after primary immunization. The survival rate was analysed 1 h after antigen challenge. On the other hand, passive systemic anaphylaxis was assessed by blood pressure monitoring. Twenty-four hours after the purified IgG fraction of anti-OVA serum (5 mg/mouse) or PBS (control) was administered intraperitoneally to wild-type and CD40-deficient mice, they were anaesthetized with an i.p. injection of pentobarbital sodium (Nembutal; Abbott Labs, North Chicago, IL) (50 μg/g) and cannulated with polyethylene tubes in the carotid artery for blood pressure monitoring and in the right jugular vein for antigen injection [13]. Blood pressure was measured by blood pressure amplifier AP-641G (Nihon Kohden, Tokyo, Japan) with Uniflow (Baxter, Irvine, CA) as the transducer. Then the mice were challenged with OVA. Blood pressure was monitored for > 1 h after OVA injection unless the mice died earlier. For measurement of plasma histamine, blood was collected for plasma preparation into EDTA tubes on ice before and 2 min after i.v. injection of OVA. Concentrations of plasma histamine were quantified by the radioimmunoassay kit (Mitsubishi Kagaku BCL, Tokyo, Japan) following the manufacturer's instructions.
Blocking of FcγR function
For blocking experiments of FcγR function, the anti-murine FcγRII/FcγRIII MoAb (2.4G2; PharMingen) was used [14,15]. The purified IgG fraction of anti-OVA serum (0.4 μg per site) was injected to CD40-deficient mice intradermally with or without 0.5 μg of 2.4G2 MoAb for PCA experiments. Standard murine IgG (Zymed Labs, South San Francisco, CA) was used as control. For passive systemic anaphylaxis, the 2.4G2 MoAb (8 μg/g body weight) was administered intraperitoneally to CD40-deficient mice 90 min before passive immunization with the purified IgG (5 mg/mouse) [15].
RESULTS
Systemic anaphylaxis in CD40-deficient mice
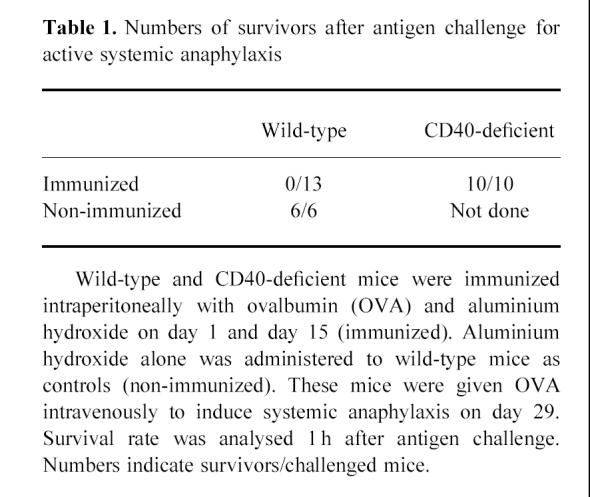
Wild-type and CD40-deficient mice were immunized with OVA and aluminium hydroxide on day 1 and day 15. Serum anti-OVA immunoglobulin levels evaluated by ELISA demonstrated the absence of humoral response other than IgM in CD40-deficient mice, whereas the increase of all classes of immunoglobulin was detected in wild-type mice (Fig. 1). Then we used these mice for the assessment of immediate systemic anaphylaxis. Intravenous administration of OVA on day 29 clearly revealed immediate systemic anaphylaxis in wild-type mice. All of them died, typically within 60 min. However, anaphylaxis was not observed in CD40-deficient mice, and all of them survived (Table 1). These results demonstrate that systemic anaphylaxis by active immunization did not take place in CD40-deficient mice, and the results indicate that the immune responses of IgG or IgA might be necessary for IgE-independent systemic anaphylaxis by active immunization in vivo.
Figure 1.
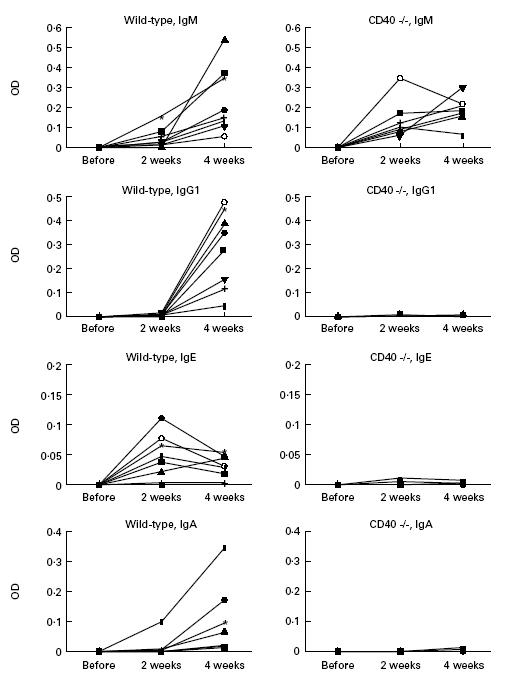
Antibody responses of wild-type and CD40-deficient mice to ovalbumin (OVA) antigen. Wild-type and CD40-deficient mice were immunized with OVA and aluminium hydroxide intraperitoneally on day 1, and boosted on day 15. Serum levels of anti-OVA antibodies for IgG1, IgG2a, IgG2b, IgG3, IgM, IgA and IgE were evaluated in each serum collected on days 1, 15 and 29 by ELISA, as described in Materials and Methods. The numbers of evaluated mice were eight for wild-type mice and seven for CD40-deficient mice. Results were expressed by the differences of optical density (OD) from the baseline, that was defined as the OD of serum obtained before immunization in each mouse. The results of IgG2a, IgG2b and IgG3 had the same tendency as IgG1, and the control group revealed no responses in all subclasses (data not shown).
Table 1.
Numbers of survivors after antigen challenge for active systemic anaphylaxis
IgG-mediated hypersensitivity reaction in CD40-deficient mice
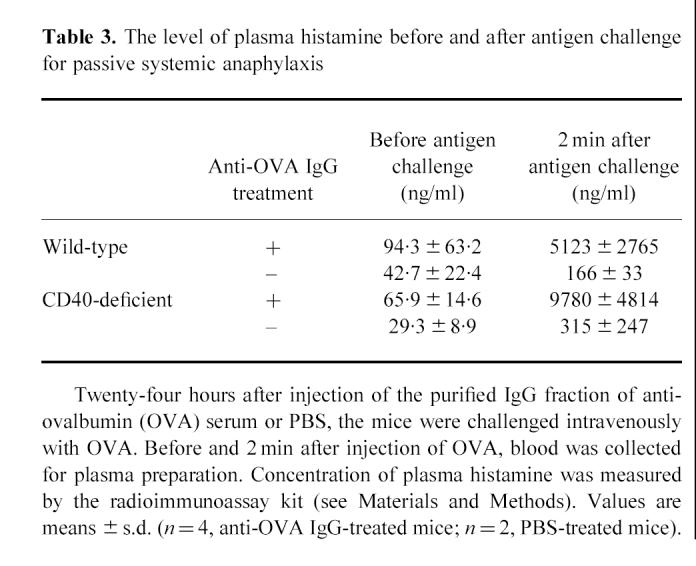
To analyse the possible role of immunoglobulin classes, especially IgG, in systemic anaphylaxis, we purified the IgG fraction from the serum of wild-type mice immunized with OVA. First, we adopted the PCA system. The purified IgG fraction of anti-OVA serum was injected intradermally on the backs of wild-type and CD40-deficient mice. OVA and Evans blue dye were injected intravenously 1 h later and the titre of PCA was determined 30 min after i.v. OVA challenge (Table 2). We obtained the positive responses of PCA in both wild-type and CD40-deficient mice when these mice were treated with the purified anti-OVA IgG but not with control IgG. Then we evaluated IgG-mediated passive systemic anaphylaxis by blood pressure monitoring system. We preliminarily observed systemic anaphylaxis both in wild-type and CD40-deficient mice by passive immunization of anti-OVA serum which contained anti-OVA IgE and IgA as well as anti-OVA IgG (data not shown). To elucidate the role of anti-OVA IgG more precisely, the blood pressure of wild-type and CD40-deficient mice passively sensitized with the purified IgG fraction of anti-OVA serum (5 mg/mouse) was monitored by cervical artery catheterization technique [13]. Following i.v. administration of OVA, blood pressure increased abruptly but then decreased rapidly within 10 min both in wild-type mice and in CD40-deficient mice (Fig. 2a,b). In addition, a marked increase in plasma histamine was observed both in wild-type and in CD40-deficient mice after antigen challenge (Table 3). The severity of anaphylaxis, that sometimes resulted in death in CD40-deficient mice passively sensitized with the purified IgG, was not different from that in wild-type mice treated similarly. In contrast, control mice treated with PBS revealed no change of blood pressure or plasma histamine level after antigen challenge (Fig. 2c and Table 3). These findings suggest that IgG might be responsible for the induction of immediate systemic hypersensitivity reaction without the humoral response of IgE.
Table 2.
Geometric mean and range of passive cutaneous anaphylaxis (PCA) titres in wild-type and CD40-deficient mice
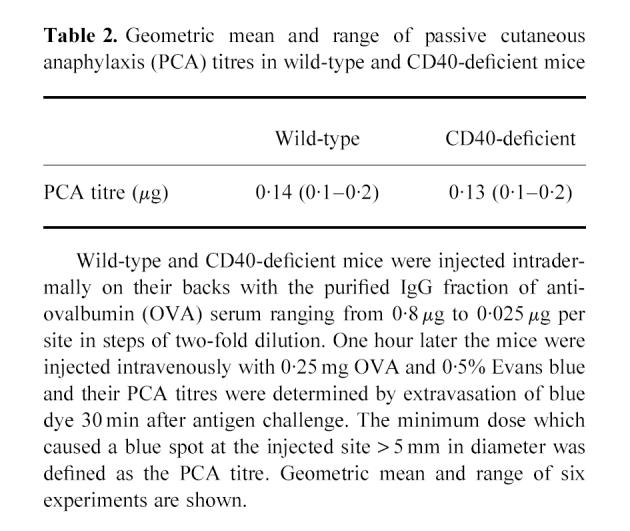
Figure 2.
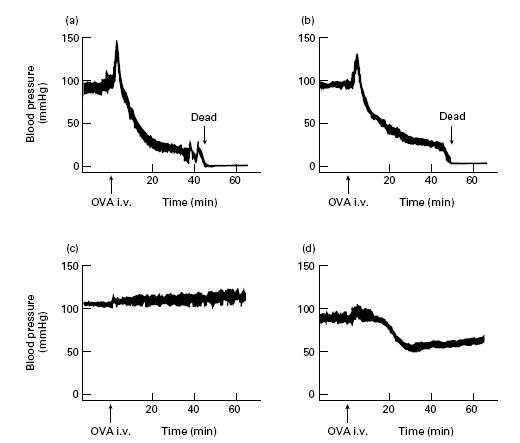
Blood pressure monitoring for passive systemic anaphylaxis. Twenty-four hours after the purified IgG fraction of anti-ovalbumin (OVA) serum injection, the mice were challenged intravenously with OVA. Blood pressure was monitored for more than 1 h after OVA injection. For the blocking of FcγR function, 2.4G2 MoAb (8 μg/g body weight) was administered intraperitoneally to CD40-deficient mice 90 min before passive immunization with the purified IgG fraction. (a) Wild-type mouse treated with the purified IgG of anti-OVA serum. (b) CD40-deficient mouse treated with the purified IgG of anti-OVA serum. (c) CD40-deficient mouse treated with PBS (control). (d) CD40-deficient mouse treated with 2.4G2 MoAb and the purified IgG of anti-OVA serum. Three independent experiments were performed with similar results. Representative data are shown.
Table 3.
The level of plasma histamine before and after antigen challenge for passive systemic anaphylaxis
Role of FcγR in systemic anaphylaxis
To investigate the inflammatory pathway of IgG-mediated hypersensitivity reactions, we focused on the FcγR function in vivo. As the anti-FcγRII/FcγRIII MoAb (2.4G2; PharMingen) has been reported to block the FcγRII/FcγRIII function [14,15], we adopted this antibody for the inhibition of both IgG-mediated PCA and IgG-mediated passive systemic anaphylaxis. The results showed that the pretreatment of the anti-FcγRII/FcγRIII antibody (0.5 μg) completely inhibited IgG-mediated PCA (Fig. 3; upper right versus upper left). Furthermore, as shown in Fig. 2d, blood pressure of CD40-deficient mice pretreated with the anti-FcγRII/FcγRIII antibody fell slowly following antigen challenge and maintained > 60 mmHg. The mice recovered within 2 h after antigen challenge and survived. Thus, we observed that IgG-mediated immediate systemic anaphylaxis assessed by blood pressure monitoring was inhibited by the i.p. administration of anti-FcγRII/FcγRIII antibody. These findings suggest that IgG initiates the immediate hypersensitivity reactions through FcγR even in in vivo conditions.
Figure 3.
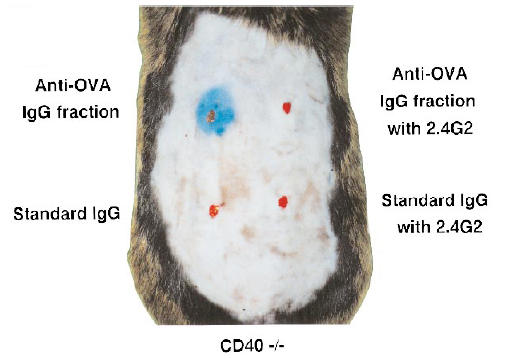
Inhibition of IgG-mediated passive cutaneous anaphylaxis (PCA) by the blocking of FcγR function in CD40-deficient mice. The purified IgG fraction of anti-ovalbumin (OVA) serum was injected into CD40-deficient mice intradermally with or without 2.4G2 MoAb. Standard murine IgG was used as control. Similar results were confirmed in four independent experiments. Upper right, anti-OVA IgG with 2.4G2; upper left, anti-OVA IgG alone; lower right, standard murine IgG with 2.4G2; lower left, standard murine IgG alone.
DISCUSSION
CD40 is a member of the tumour necrosis factor receptor family of cell surface proteins and has been shown to be an essential molecule of B cell class switching for T cell-dependent antigens [6]. In animal models, CD40-deficient mice lack the immunoglobulin class switching for T cell-dependent antigens and their immune responses have been shown to be quite similar to those of X-linked hyper-IgM syndrome, which is caused by human genetic disorder of CD40 ligand. IgG, IgA and IgE responses, but not the IgM response, are not induced by immunization of T cell-dependent antigens both in CD40-deficient mice and patients with X-linked hyper-IgM syndrome. Thus, CD40-deficient mice are useful models for analysing the role of immunoglobulin classes on immunological reactions. We show in this study that systemic anaphylaxis did not take place in CD40-deficient mice by the active immunization of T cell-dependent antigens. In contrast to CD40-deficient mice, IgE-deficient mice sensitized with antigen revealed anaphylaxis on antigen challenge [5]. Considered with the other findings from in vitro experiments suggesting that non-IgE stimuli might cause anaphylaxis [2–4,16,17], we hypothesized that IgG, which is a homocytotropic antibody other than IgE, might be responsible for the immediate systemic anaphylaxis. In this study we demonstrate the positive responses of PCA and systemic anaphylaxis by transferring the antigen-specific IgG fraction both in CD40-deficient mice and wild-type mice. Taking all the data into consideration, we propose that IgG plays an important role in triggering IgE-independent hypersensitivity reactions even in in vivo conditions. In human beings the contribution of IgG antibodies to anaphylaxis remains a topic of discussion, and it may not be easy to adopt our results into human beings directly. However, as some investigators recently reported an anaphylactic role of IgG [18,19], further investigation would be required.
The FcR γ subunit is an essential component of not only the high-affinity receptor for IgE (FcɛRI), but also the high- and low-affinity receptors for IgG (FcγRI and FcγRIII) [20,21]. Targeted disruption of the FcR γ subunit gene in mice resulted in the absence of IgE-mediated anaphylaxis and Arthus reaction [9,22]. Although FcγRI is mainly expressed on phagocytes and not on mast cells, FcγRIII is present on murine mast cells [23]. FcγRII, the other low-affinity receptor for IgG, is also expressed on mast cells, but it acts as a general negative regulator through phosphorylated immunoreceptor tyrosine-based inhibition motif, which inhibits FcɛRI-mediated or immune complex-triggered activation [12,24,25]. In the light of these findings and our results, we hypothesized that the blocking of FcγRIII may reduce the IgG-mediated immediate hypersensitivity reactions. In fact, we demonstrated that pretreatment with 2.4G2, anti-murine FcγRII/FcγRIII MoAb, resulted in the inhibition of IgG-mediated systemic anaphylaxis in in vivo conditions. Taken together with other investigations [16,17,26–29], we speculated that IgG-mediated anaphylaxis was triggered through FcγRIII, and the blocking of FcγR would provide us with one of therapeutic targets for the control of immediate hypersensitivity diseases.
Recently, Miyajima et el. and Dombrowicz et al. reported that IgG could induce anaphylaxis via FcγRIII, which was similar to our results [26,27]. In addition to the essential role of FcγRIII in the induction of anaphylaxis, they indicated the important role of cell populations other than mast cells. However, histamine has been considered to be derived only from mast cells or basophils in mice. We demonstrate here that plasma histamine was markedly increased after induction of anaphylaxis, suggesting that mast cells play an essential role even in IgG-mediated anaphylaxis.
Again, although the IgE-triggered release of mast cell mediators in response to antigen is thought to be the primary event in immediate hypersensitivity reactions such as systemic anaphylaxis, previous reports showed that mast cells can be activated by IgE-independent mechanisms [2–5]. Furthermore, mast cells have been shown to express FcγR [23]. Taking all these data together, we speculated that the interaction of antigen, antigen-specific IgG, and its cognate FcγR on the cell surface of mast cells is another step triggering an immediate hypersensitivity reaction and anaphylaxis (type I hypersensitivity reaction). Recently, Ravetch reported the important role of FcγR in initiating IgG-mediated inflammatory reactions [10]. Genetic deletion of the γ subunit of Fc receptors in mice resulted in the attenuation of the cytotoxic antibody responses (type II hypersensitivity reaction) [8] and the Arthus reaction triggered by immune complexes did not occur in the absence of FcγR even if an intact complement system was present (type III hypersensitivity reaction) [9,28,29]. Taking these findings and our results into consideration, we suggest that the antigen–IgG complex directly bound to FcγRIII on mast cells stimulates their degranulation, and FcγRIII may play the key role in triggering the IgE-independent immediate hypersensitivity reaction as well as the other IgG-mediated inflammatory reactions such as type II and type III hypersensitivity reactions. In addition to the classical immunological dogma of anaphylaxis in IgE-triggering type I hypersensitivity, the IgG-triggering type I hypersensitivity should be considered. Furthermore, we speculate that FcγR may be one of the common targets for the treatment of allergic diseases including IgG-mediated hypersensitivity reactions of type I, II and III.
Acknowledgments
We thank Y. Yamashita for his advice on IgG purification and T. Hasegawa and L. Wang for the experiments on mouse blood pressure monitoring.
REFERENCES
- 1.Dombrowicz D, Flamand V, Brigman KK, Koller BH, Kinet J-P. Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor α chain gene. Cell. 1993;75:969–76. doi: 10.1016/0092-8674(93)90540-7. [DOI] [PubMed] [Google Scholar]
- 2.Daëron M, Couderc J, Ventura M, Liacopoulos P, Voisin GA. Anaphylactic properties of mouse monoclonal IgG2a antibodies. Cell Immunol. 1982;70:27–40. doi: 10.1016/0008-8749(82)90130-7. [DOI] [PubMed] [Google Scholar]
- 3.Fox PC, Basciano LK, Siraganian RP. Mouse mast cell activation and desensitization for immune aggregate-induced histamine release. J Immunol. 1982;129:314–9. [PubMed] [Google Scholar]
- 4.Katz HR, Raizman MB, Gartner CS, Scott HC, Benson AC, Austen KF. Secretory granule mediator release and generation of oxidative metabolites of arachidonic acid via Fc-IgG receptor bridging in mouse mast cells. J Immunol. 1992;148:868–71. [PubMed] [Google Scholar]
- 5.Oettgen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, Leder P. Active anaphylaxis in IgE-deficient mice. Nature. 1994;370:367–70. doi: 10.1038/370367a0. [DOI] [PubMed] [Google Scholar]
- 6.Kawabe T, Naka T, Yoshida K, et al. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1:167–78. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 7.Coombs RRA, Gell PGH. Classification of allergic reactions responsible for clinical hypersensitivity and disease. In: Gell PGH, Coombs RRA, Lachmann PJ, editors. Clinical aspects of immunology. 3. Oxford: Blackwell Scientific Publications; 1975. pp. 761–81. [Google Scholar]
- 8.Clynes R, Ravetch JV. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity. 1995;3:21–26. doi: 10.1016/1074-7613(95)90155-8. [DOI] [PubMed] [Google Scholar]
- 9.Sylvestre DL, Ravetch JV. Fc receptors initiate the Arthus reaction: redefining the inflammatory cascade. Science. 1994;265:1095–8. doi: 10.1126/science.8066448. [DOI] [PubMed] [Google Scholar]
- 10.Ravetch JV. Fc receptors: rubor redux. Cell. 1994;78:553–60. doi: 10.1016/0092-8674(94)90521-5. [DOI] [PubMed] [Google Scholar]
- 11.Ovary Z. Passive cutaneous anaphylaxis in the mouse. J Immunol. 1958;81:355–7. [PubMed] [Google Scholar]
- 12.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature. 1996;379:346–9. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Hasegawa T, Nadai M, Muraoka I, Nabeshima T, Kato N. The effect of lipopolysaccharide on the disposition of xanthines in rats. J Pharm Pharmacol. 1993;45:34–38. doi: 10.1111/j.2042-7158.1993.tb03675.x. [DOI] [PubMed] [Google Scholar]
- 14.Unkeles JC. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte Fc receptors. J Exp Med. 1979;150:580–96. doi: 10.1084/jem.150.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurlander RJ, Ellison DM, Hall J. The blockade of Fc receptor-mediated clearance of immune complexes in vivo by a monoclonal antibody (2.4G2) directed against Fc receptors on murine leukocytes. J Immunol. 1984;133:855–62. [PubMed] [Google Scholar]
- 16.Daëron M, Bonnerot C, Latour S, Fridman WH. Murine recombinant FcγRIII, but not FcγRII, trigger serotonin release in rat basophilic leukemia cells. J Immunol. 1992;149:1365–73. [PubMed] [Google Scholar]
- 17.Latour S, Bonnerot C, Fridman WH, Daëron M. Induction of tumor necrosis factor-α production by mast cells via FcγR. Role of the FcγRIII γ subunit. J Immunol. 1992;149:2155–62. [PubMed] [Google Scholar]
- 18.Jimeno L, Lombardero M, Carreira J, Moscoso del Prado J. Presence of IgG4 on the membrane of human basophils. Histamine release is induced by monoclonal antibodies directed against the Fab but not the Fc region of IgG4 molecule. Clin Exp Allergy. 1992;22:1007–14. doi: 10.1111/j.1365-2222.1992.tb03029.x. [DOI] [PubMed] [Google Scholar]
- 19.Schuurman J, Perdok GJ, Mueller GA, Aalberse RC. Complementation of Der p 2-induced histamine release from human basophils sensitized with monoclonal IgE: not only by IgE, but also by IgG antibodies directed to a nonoverlapping epitope of Der p 2. J Allergy Clin Immunol. 1998;101:404–9. doi: 10.1016/S0091-6749(98)70255-6. [DOI] [PubMed] [Google Scholar]
- 20.Ra C, Jouvin M-HE, Blank U, Kinet J-P. A macrophage Fcγ receptor and the mast cell receptor for IgE share an identical subunit. Nature. 1989;341:752–4. doi: 10.1038/341752a0. [DOI] [PubMed] [Google Scholar]
- 21.Kurosaki T, Ravetch JV. A single amino acid in the glycosyl phosphatidylinositol attachment domain determines the membrane topology of FcγRIII. Nature. 1989;342:805–7. doi: 10.1038/342805a0. [DOI] [PubMed] [Google Scholar]
- 22.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–29. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 23.Katz HR, Arm JP, Benson AC, Austen KF. Maturation-related changes in the expression of FcγRII and FcγRIII on mouse mast cells derived in vitro and in vivo. J Immunol. 1990;145:3412–7. [PubMed] [Google Scholar]
- 24.Daëron M, Malbec O, Latour S, Arock M, Fridman WH. Regulation of high-affinity IgE receptor-mediated mast cell activation by murine low-affinity IgG receptors. J Clin Invest. 1995;95:577–85. doi: 10.1172/JCI117701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malbec O, Fong DC, Turner M, Tybulewicz VLJ, Cambier JC, Fridman WH, Daëron M. Fcɛ receptor I-associated lyn-dependent phosphorylation of Fcγ receptor IIB during negative regulation of mast cell activation. J Immunol. 1998;160:1647–58. [PubMed] [Google Scholar]
- 26.Miyajima I, Dombrowicz D, Martin TR, Ravetch JV, Kinet J-P, Galli SJ. Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and FcγRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J Clin Invest. 1997;99:901–14. doi: 10.1172/JCI119255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dombrowicz D, Flamand V, Miyajima I, Ravetch JV, Galli SJ, Kinet JP. Absence of FcɛRI α chain results in upregulation of FcγRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between FcɛRI and FcγRIII for limiting amounts of FcR β and γ chains. J Clin Invest. 1997;99:915–25. doi: 10.1172/JCI119256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hazenbos WLW, Gessner JE, Hofhuis FMA, et al. Impaired IgG-dependent anaphylaxis and Arthus reaction in FcγRIII (CD16) deficient mice. Immunity. 1996;5:181–8. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 29.Sylvestre DL, Ravetch JV. A dominant role for mast cell Fc receptors in the Arthus reaction. Immunity. 1996;5:387–90. doi: 10.1016/s1074-7613(00)80264-2. [DOI] [PubMed] [Google Scholar]