

Abstract
ANCA directed against PR3 are highly specific for Wegener's granulomatosis and microscopic polyangiitis, and have been implicated in the pathogenesis of small vessel vasculitis. Most PR3-ANCA are directed against conformational epitopes on PR3. This study was designed to determine whether the cleavage of the N-terminal activation dipeptide of PR3 is required for the binding of PR3-ANCA. Recombinant PR3 (rPR3) variants were expressed in the epithelial cell line, 293. As confirmed by radiosequencing, the rPR3 secreted into the 293 cell culture supernatant is N-terminally unprocessed. Two enzymatically inactive rPR3 mutants were expressed in 293 cells: rPR3-S176A and δ-rPR3-S176A. rPR3-S176A contains the N-propetide Ala-2-Glu-1, δ-rPR3-S176A does not. Culture supernatants of rPR3-S176A and δ-rPR3-S176A expressing 293 cells were used as sources of target antigen for PR3-ANCA testing by capture ELISA. Forty unselected consecutive PR3-ANCA+ sera were tested. With δ-rPR3-S176A as antigen all 40 were recognized, compared with only 34 of 40 when rPR3-S176A served as target antigen. The majority of the serum samples contained a mixture of antibodies reacting with epitopes accessible on the mature and on the proform of PR3. In conclusion, the cleavage of the N-terminal activation dipeptide of PR3 is not an absolute requirement for recognition by all PR3-ANCA. However, a substantial proportion of PR3-ANCA recognize (a) target antigen(s) exposed only after the conformational change of PR3 associated with the N-terminal processing. In 15% of sera this PR3-ANCA subset occurred exclusively. PR3-ANCA subtypes can be differentiated using specifically designed rPR3 variants as target antigens, and non-haematopoietic mammalian cells without regulated secretory pathway can be used for their expression.
Keywords: anti-neutrophil cytoplasmic antibodies, target antigen, recombinant proteinase 3, N-terminal propeptide
INTRODUCTION
ANCA yielding a characteristic cytoplasmic immunofluorescence pattern on ethanol-fixed neutrophils (c-ANCA) occur in most patients with active Wegener's granulomatosis (WG) and a subset of patients with microscopic polyangiitis [1,2]. The neutral serine protease, PR3, which is a constituent of azurophil granules of neutrophils, has been identified as the principal target antigen for c-ANCA in WG [3–5]. Competition analysis using MoAbs indicates that ANCA directed against PR3 (PR3-ANCA) from patients with WG recognize several different epitopes on the PR3 molecule [6]. The majority of PR3-ANCA appear to be directed against conformational epitopes of PR3 [7].
Recently, different expression systems have been used to express recombinant PR3 (rPR3) variants [8–12]. Full length cDNA constructs encoding the PR3 zymogen cloned into baculovirus vectors have been expressed in Sf9 insect cells [8–10]. It has been reported that the N-terminal activation dipeptide needs to be removed in vitro from the expressed protein in order to obtain the active enzyme with the N-terminal amino acid sequence Ile-Val-Gly-Gly [10]. The N-terminally unprocessed rPR3 zymogen from Sf9 cells is recognized only by a small number of PR3-ANCA-containing sera [8,9]. In contrast, enzymatically active, N-terminally processed rPR3 expressed in the human mast cell line, HMC-1, appears to be recognized by all PR3-ANCA [13,14]. Based on these observations, we hypothesized that the conformational change associated with the cleavage of the N-terminal propeptide of PR3 is required for recognition by most PR3-ANCA.
To test this hypothesis, we used N-terminally processed and N-terminally unprocessed rPR3 as novel target antigens in a previously described capture ELISA [14] for PR3-ANCA testing. The findings presented here indicate that a substantial proportion of PR3-ANCA react with epitopes on PR3 that are only accessible after cleavage of the N-terminal propeptide. Some patients appear to only have PR3-ANCA reacting with these epitopes. In contrast, we did not detect patients that had PR3-ANCA reacting exclusively with the proform of PR3.
MATERIALS AND METHODS
Materials
Unless specified otherwise, all reagents were purchased from Sigma (St Louis, MO).
Cell culture
The HMC-1/PR3-S176A cell line expressing the inactive mutant rPR3-S176A used for PR3-ANCA testing by indirect immunofluorescence (IIF), and the human fetal kidney epithelial cell line, 293, obtained from ATCC (Rockville, MD), were cultured as described previously [11,14,15].
cDNA constructs, expression vectors and transfection procedures
A scheme of the various rPR3 c-DNA constructs used in this study is shown in Fig. 1. The original cDNA insert of wild-type PR3 spanning from nucleotide positions 9 to 790 of the published sequence [16] as well as the cDNA insert coding for the inactive mutant rPR3-S176A were prepared as described elsewhere [11]. The deletion mutants lacking the codons for the N-terminal propeptide (δ-rPR3 and δ-rPR3-S176A) were prepared using the splicing by overlap extension method [17] with the primers listed in Table 1. To generate the cDNA construct coding for δ-rPR3, overlapping DNA fragments were prepared by polymerase chain reaction (PCR) using primers 7 and 4, and primers 8 and 3, respectively, with wild-type PR3 cDNA as template. The resulting DNA fragments were spliced together and cloned into the expression vector pRcCMV (Invitrogen, San Diego, CA). Similarly, to generate the cDNA construct coding for δ-rPR3-S176A, the Δ-PR3 cDNA was used as template with primers 5 and 4, and primers 6 and 3, respectively. The resulting expression plasmid was designated pRcCMV/ΔPR3-S176A.
Fig.1.

PR3 cDNA constructs. The constructs carrying the mutation S176A have an alanine residue in position 176 of the published amino acid sequence [16] instead of the active site serine. This mutation results in lack of enzyme activity [11] but does not affect recognition by PR3-ANCA [14]. The constructs δ-rPR3 and δ-rPR3-S176A do not code for the N-terminal propeptide, resulting in expression of mature rPR3 with an isoleucine residue in the first position.
Table 1.
Primers used in the preparation of the cDNA insert coding for δ-rPR3-S176A

The calcium-phosphate precipitation method was used for transfection of adherent 293 cells as described elsewhere [15]. Transfected 293 cell clones selected in the presence of 700 μg/ml G418 were screened for rPR3 expression by IIF using a rabbit polyclonal anti-PR3 serum and the MoAb MCPR3-2 [14].
Immunologic methods
The MoAb MCPR3-2 and the polyclonal anti-PR3 rabbit serum have been described elsewhere [14]. IIF was performed on ethanol-fixed cytospin preparations as described [11].
The capture ELISA used for the detection and quantification of rPR3 in 293 cell culture supernatants with MCPR3-2 as capturing antibody and the rabbit polyclonal anti-PR3 serum as detecting antibody has been described in detail elsewhere [14]. The same assay was also used for PR3-ANCA detection as previously described [14], only with the different novel rPR3 target antigens. For the studies presented here serum-free culture supernatants of 293 cell clones expressing either δ-rPR3-S176A or rPR3-S176A served as antigen sources. They were prepared by washing the cell monolayers once with serum-free medium, followed by incubation with fresh serum-free medium for 48 h. Saturation of the binding capacity of MCPR3-2 adsorbed to plastic wells, as determined using the rabbit anti-PR3 serum for detection, was achieved with a 1:32 dilution of δ-rPR3-S176A and a 1:64 dilution of rPR3-S176A-containing media, respectively. Subsequently, 200 μl of diluted antigen-containing medium were incubated in MCPR3-2-coated wells for 1 h at room temperature. MCPR3-2-coated wells incubated with antigen-free medium served as control. Based on studies including large numbers of sera from both normal and disease control patients, a net absorbance of < 0.100 is considered negative in this assay [14].
For immunoblotting, proteins contained in 50-μl aliquots of serum-free culture supernatants obtained after 48 h of incubation on confluent monolayers (2 ml of media1/.5 × 107 cells) of rPR3-S176A and δ-rPR3-S176A-expressing cells were separated by SDS–PAGE (12% gels) under non-reducing conditions, transferred to Immun-Lite membrane (BioRad, Hercules, CA). The filters were probed either with the MoAb MCPR3-2 or with irrelevant monoclonal mouse control IgG1. Bound antibody was visualized using the Immun-Lite chemiluminescence detection system according to the manufacturer's instructions (BioRad).
Metabolic labelling and radiosequencing
For pulse-chase experiments, cells grown to confluence were washed once and starved for 30 min in methionine- and cysteine-free culture medium/5% bovine calf serum (BCS). Cells were then incubated for 30 min in the same media supplemented with 100 μCi/ml 35S-methionine/35S-cysteine (pulse labelling). Subsequently, the cells were washed once, resuspended and cultured in regular culture medium/10% BCS for the various time periods (chase). Ten million cells were collected and centrifuged for each time point. The cell pellets and the supernatants were snap frozen separately, and stored at −20°C until analysis by immunoprecipitation, SDS–PAGE, and autoradiography.
Cell supernatants from 10 × 106 cells were normalized to 6 × 106 ct/min by trichloroacetic acid (TCA) precipitation, and precleared by incubation with 50 μl of Staphylococcus aureus (Gibco, Gaithersburg, MD) for 30 min at 4°C, followed by centrifugation at 10 000 g for 2 min. The supernatants were incubated with 5 μl of antibody at 4°C overnight. Staphylococcus aureus (50 μl) was again added and incubated at 4°C for 1 h. Subsequently the immunoprecipitates were washed three times in 750 μl of radioimmune precipitation buffer (50 mm Tris–HCl, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 50 mm NaF, 100 μm Na-vanadate, 75 μg/ml PMSF, 0.1 trypsin inhibitory units (TIU) aprotinin in PBS, pH 8.0), dissolved in 80 μl of SDS–PAGE buffer (0.4 m Tris–HCl, 50% (v/v) glycerol, 10% (w/v) SDS, 5% (v/v) mercaptoethanol), boiled and electrophoresed on 12% SDS–PAGE gels. After fixation the gels were washed in 2,5-diphenyloxazole enhancing solution (Fisher Scientific, Fair Lawn, NJ) for 90 min, dried onto filter paper, and exposed to Kodak XAR-5 film (Kodak, Rochester, NY) for various time periods at −70°C.
For radiosequencing, the above procedure was modified as follows. For each time point 6 × 107 cells were washed and starved for 30 min in methionine- and isoleucine-free culture medium/5% BCS. Cells were then pulse-labelled for 30 min in the same media supplemented with 200 μCi/ml 35S-methionine and 200 μCi/ml 3H-isoleucine. After the various chase time periods media supernatants were collected and snap frozen. Immunoprecipitated proteins were separated by SDS–PAGE, transferred to PVDF membrane (BioRad) and visualized by exposure to Kodak XAR-5 film (Kodak) overnight. The exposed film served as template for identification of the protein bands that were cut out of the PVDF membrane and subjected to N-terminal sequence analysis using an Applied Biosystems 470A sequencer. The resulting phenylthiohydantoin amino acids were assayed for tritium using standard scintillation counting procedures.
Deglycosylation of immunoprecipitates
To determine the presence of N-linked glycosylation, rPR3 was subjected to digestion with N-glycosydase F. Immunoprecipitates of culture supernatants from metabolically labelled cells were prepared, washed, resuspended in 10 μl of 0.5% SDS, and incubated for 2 min at 95°C. After addition of 90 μl of digestion buffer (20 mm Na2HPO4, 10 mm NaN3, 50 mm EDTA, 0.5% (v/v) Nonidet P-40, pH 7.2), the sample was divided in two. One sample served as the control; 0.4 U N-glycosidase F (Boehringer, Mannheim, Germany) was added to the second sample, and both were incubated at 37°C for 15 h. The samples were then precipitated in 30% TCA, washed in cold acetone and analysed by SDS–PAGE and autoradiography.
Patient sera
The assays were performed using 40 consecutive PR3-ANCA+ serum samples from individual patients obtained from the Clinical Immunology Laboratory (Mayo Clinic, Rochester, MN). No patient identifiers or clinical data about these patients were available to the investigators.
RESULTS
Expression of rPR3 variants in 293 cells
The fetal kidney epithelial cell line, 293, only has a non-regulated secretory pathway, resulting in secretion of the majority of newly synthesized recombinant proteins into the culture media supernatants. We transfected 293 cells with the expression plasmids pRcCMV/δPR3-S176A and pRcCMV/PR3-S176A [11] coding for δ-rPR3-S176A lacking the N-terminal propeptide, and rPR3-S176A, respectively. To avoid cell toxicity resulting from prematurely activated rPR3, we used the rPR3 variants containing the mutation Ser-176 to Ala-176 which deprives the molecule of its enzymatic activity [11] but does not affect the binding of PR3-ANCA [14].
The rPR3 variants secreted into the 293 cell culture media were analysed by PR3-specific capture ELISA (Fig. 2) [14], immunoblotting (Fig. 3a) and immunoprecipitation (Fig. 3b,c). As determined by capture ELISA, 1.1 μg/ml of rPR3-S176A and 0.55 μg/ml of δ-rPR3-S176A were secreted into the culture media. The slopes of the linear portions of the antigen dilution curves determined by capture ELISA are parallel, suggesting that the affinity of the two rPR3 variants for the capturing MoAb MCPR3-2 is similar (Fig. 2). The rPR3 proteins secreted into the media have an approximate molecular mass of 32–38 kD (Fig. 3). This molecular mass is equivalent to the unprocessed large molecular variant of rPR3 secreted into the culture supernatants of HMC-1/PR3-S176A cells (Fig. 3c) and of U937 cells [11]. After removal of the asparagine-linked sugar moeities by treatment of rPR3 with N-glycosidase F the molecular mass was reduced to about 29 kD (Fig. 3c). This indicates that, as previously observed for U937 and HMC-1/PR3 cells, the large unprocessed rPR3 variant is secreted into the media of 293 cells.
Fig. 2.
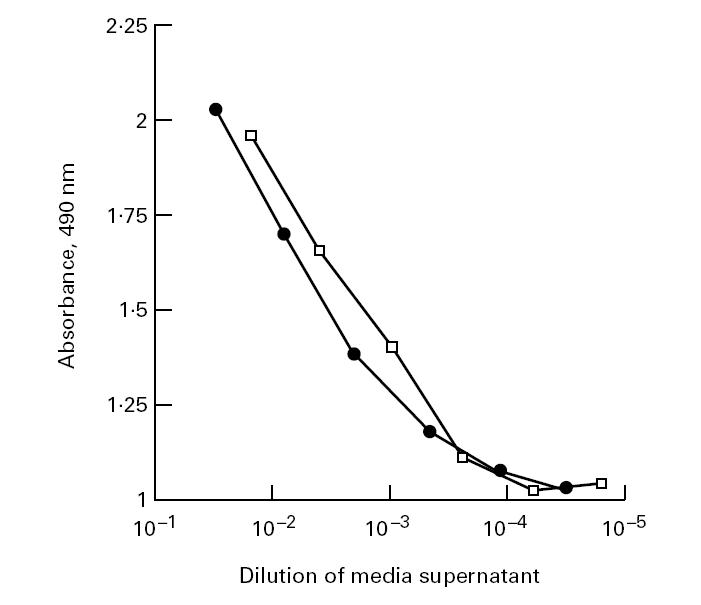
Detection of rPR3 variants secreted into the media supernatants of stably transfected 293 cells by capture ELISA. Shown are dilution profiles of serum-free media culture supernatants of 293 cell clones expressing rPR3-S176A (□) and δ-rPR3-S176A (•). The MoAb MCPR3-2 was used as capturing antibody, and the rabbit polyclonal anti-PR3 serum was used as detecting antibody in the assay [14].
Fig. 3.

Characterization of rPR3 secreted into the media supernatants of stably transfected 293 cells. (a) Immunoblot of proteins contained in media supernatants of 293 cells expressing rPR3-S176A (lane 1) and δ-rPR3-S176A (lane 2) separated by SDS–PAGE (12% gel, non-reducing conditions). Detection of the rPR3 variants with the PR3-specific MoAb MCPR3-2 reveals an electrophoretic mobility consistent with a molecular mass of about 38 kD. (b) 293 cells expressing rPR3-S176A were pulse-labelled with 35S-methionine/35S-cysteine and subsequently incubated with normal growth medium for the time periods (hours of chase) indicated at the bottom of the panel. Labelled proteins secreted into the media supernatants were immunoprecipitated with the rabbit anti-PR3 serum and separated by SDS–PAGE (12% gel) under reducing conditions. The bulk of the newly synthesized rPR3 was secreted into the media within 90 min. The majority of the immunoprecipitated material has an apparent molecular mass of 34–36 kD (line). The minor immunoprecipitated 32-kD band (arrow) was also an N-terminally unprocessed rPR3-S176A isoform (confirmed by radiosequencing). (c) Media supernatants from metabolically labelled HMC-1/PR3-S176A cells and from 293 cells expressing rPR3-S176A were immunoprecipitated with the rabbit anti-PR3 serum and divided into two equal aliquots each, which were incubated with (+) or without (–) N-glycosidase F under identical conditions followed by SDS–PAGE (12% gel, reducing conditions) and autoradiography. N-glycosidase F digestion resulted in removal of all N-linked sugar moeities and a drop of the molecular mass to 29 kD. Comparison of rPR3-S176A expressed in HMC-1 cells (lanes 1 and 2) to that expressed in 293 cells (lanes 3 and 4) indicates that the glycosylation of the rPR3 secreted into the media supernatants of these two cell types is similar.
Radiosequencing of immunoprecipitated rPR3-S176A confirmed that the N-propetide was not cleaved of rPR3-S176A secreted into the media of 293 cells (Fig. 4a). In contrast, the cells stably transfected with the plasmid coding for δ-rPR3-S176A secreted rPR3 with isoleucine in the first position into their media (Fig. 4b).
Fig. 4.
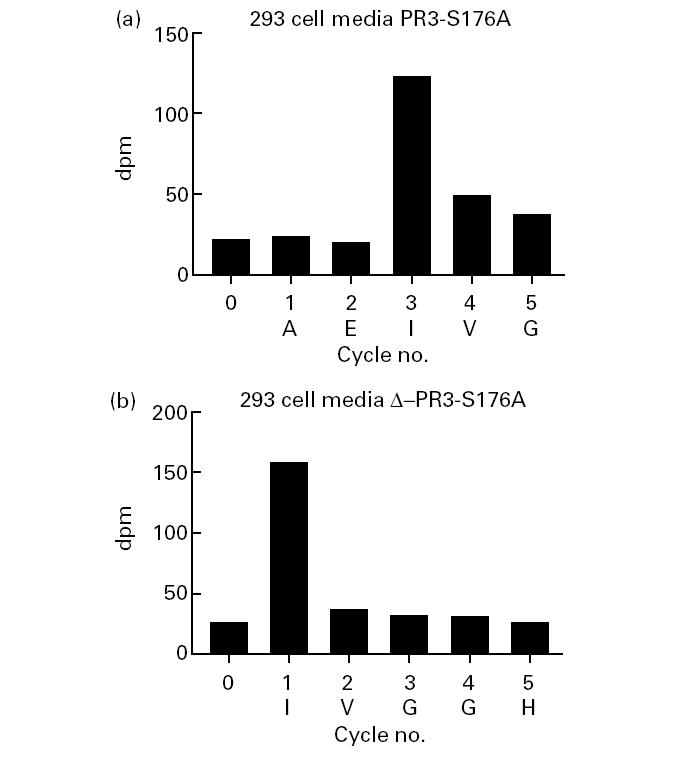
Protein sequencing of 3H-isoleucine-labelled rPR3 variants secreted into 293 cell media supernatants. Biosynthetically labelled proteins were immunoprecipitated with the rabbit anti-PR3 serum, separated by SDS–PAGE (12% gel), transferred to PVDF-membrane. The PR3-specific 32–36-kD protein bands were cut out of the filter and sequenced by Edman degradation followed by scintillation counting of radioactivity. Shown is the radioactivity (ct/min) associated with each sequence cycle. The rPR3-S176A protein secreted into 293 cell media has an isoleucine residue in the third position (a), whereas the δ-rPR3-S176A protein has isoleucine in the first position (b). The corresponding predicted N-terminal amino acid sequence (single-letter code) is shown at the bottom of each panel.
Binding of PR3-ANCA to rPR3-S176A and δ-rPR3-S176A secreted into 293 cell media
To determine whether the N-terminal proteolytic processing and the associated conformational change of PR3 affect the binding of PR3-ANCA, we tested 40 consecutive serum samples containing PR3-ANCA in the capture ELISA using either rPR3-S176A or δ-rPR3-S176A secreted into 293 cell culture supernatants as target antigen. The presence of PR3-ANCA in these sera had previously been determined by positivity in both the IIF test using HMC-1/PR3 cell cytospin preparations [13], and the capture ELISA using purified PMN-PR3 as antigen [14].
All 40 samples were recognized in the capture ELISA when δ-rPR3-S176A media were used as target antigen. Six of the 40 serum samples were negative in the assay when rPR3-S176A media were used as antigen. Figure 5 shows a scattergram of the net absorbance values obtained for each serum sample with the two different target antigens. Subsequently, serial dilutions of serum samples yielding the same net absorbance with both target antigens and of serum samples yielding different net absorbance values were assayed in parallel on both target antigens (Fig. 6). The shape of the dilution curves overlaps for sera yielding equivalent absorbance with both target antigens (Fig. 6a), indicating these sera only contain antibodies reactive with epitopes that are equally accessible on the proform and on the mature form of PR3. In contrast, the shape of the dilution curve differs for sera yielding different net absorbance on both target antigens (Fig. 6b), indicating that PR3-ANCA subsets reacting with different epitopes are present.
Fig. 5.
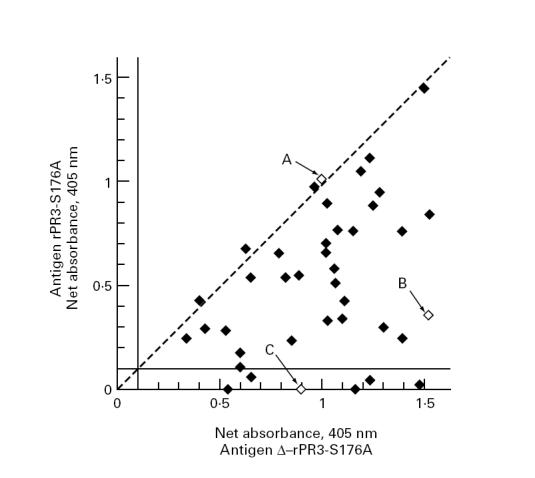
Scattergram of absorbance values obtained by capture ELISA with the two different rPR3 substrates. Forty consecutive PR3-ANCA+ serum samples were evaluated by capture ELISA using culture supernatants either from rPR3-S176A-expressing 293 cells (ordinate) or from δ-rPR3-S176A-expressing 293 cells (abscissa) as target antigen. Net absorbance values of < 0.100 are considered negative in this assay. The dashed diagonal reference line indicates equal reactivity with both target antigens. The majority of samples contained some antibodies that reacted with epitopes that only became accessible on the PR3 molecule after cleavage of the N-terminal activation dipeptide. The three serum samples marked by arrows A, B, and C are the ones for which the dilution curves are shown in Fig. 6.
Fig. 6.
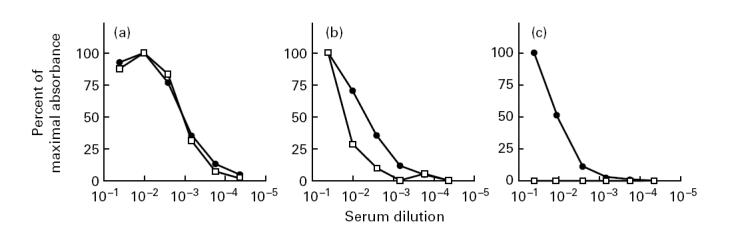
Dilution curves of three representative PR3-ANCA-containing sera. Shown are the dilution curves of the three sera indicated in Fig. 5 as A, B, and C, representing examples of three types of sera: (a) yielding equivalent absorbance values with both target antigens; (b) yielding positive, but substantially different absorbance values with the two target antigens; (c) showing no reactivity with the unprocessed proform of rPR3-S176A. The data are expressed as percentage of maximal absorbance obtained with a serum dilution of 1:20. •, Reactivity of the serum samples with δ-rPR3-S176A; □, reactivity with rPR3-S176A.
DISCUSSION
The data presented here indicate that a distinct subset of PR3-ANCA epitopes is accessible only after the conformational change associated with the cleavage of the N-terminal activation dipeptide of PR3. Most PR3-ANCA sera contain a mixture of antibodies reactive with different epitopes on the PR3 molecule. PR3-ANCA of some patients (15%) reacted exclusively with epitopes only accessible on the mature form of PR3. In contrast, no sera were detected that reacted with epitopes that are unique to the proform of PR3.
The observed different binding of antibodies from different sera to the two rPR3 variants cannot be explained by different binding of the target antigens to the plate, because the affinity of antigens to the capturing MoAb MCPR3-2 was equal, the binding of polyclonal rabbit antibody to both rPR3 variants was equal, and the sera yielding equal absorbance on both substrates had overlapping dilution curves. Different binding of α1-PI from serum to the two rPR3 variants also cannot explain the different PR3-ANCA binding. As a consequence of the S176A mutation neither rPR3 variant used could form covalent complexes with α1-PI. If different reversible complex formation with α1-PI from serum affected the binding of PR3-ANCA, less PR3-ANCA binding to δ-rPR3-S176A would be expected, as this is the variant in the conformation of the mature enzyme expected to bind more α1-PI. However, the opposite was observed.
PR3-ANCA from patients with WG appear to be directed against a few dominant epitopes [6]. Different experimental approaches have suggested that most of these epitopes are conformational [3,7], a fact which has complicated their unequivocal identification using standard methods of epitope mapping which involve linear peptide fragments [18,19]. Recently, it has been proposed that the majority of PR3-ANCA epitopes are located in flexible regions of the molecule accessible on the proform of PR3 [10,18]. The primary rPR3 protein expressed in insect cells using baculovirus expression systems is N-terminally unprocessed and needs to be activated in vitro after partial purification [10]. The only rPR3 variants expressed in insect cells that were tested for reactivity with PR3-ANCA were not recognized by the majority of PR3-ANCA+ sera [8,9]. In contrast, native PMN-PR3 or N-terminally processed rPR3 expressed in HMC-1 cells, displayed in a conformationally intact fashion, represent the most sensitive substrates for PR3-ANCA testing [13,14,20].
Our findings are consistent with these previous observations, in that all PR3-ANCA+ sera were recognized by the N-terminally processed form of rPR3, whereas only 85% of the sera also reacted with the N-terminally unprocessed proform of the enzyme. Conversely, this also means that 85% of PR3-ANCA+ sera contained some antibodies that did react with epitopes accessible on the unprocessed proform of PR3. Thus, our original hypothesis, that cleavage of the N-terminal propeptide is a requirement for recognition of PR3 by PR3-ANCA, was not supported. Furthermore, the fact that rPR3 expressed in Sf9 cells was not recognized by the majority of c-ANCA+ sera cannot fully be explained by its N-terminally unprocessed state.
It is remarkable that 15% of patients had PR3-ANCA that only reacted with epitopes on the mature form of PR3, whereas none had PR3-ANCA that reacted with epitopes accessible exclusively on the proform. This observation implies that PR3-ANCA epitopes are not lost as a consequence of the activation of PR3. Therefore, our data indicate that epitopes for PR3-ANCA are unlikely to reside on the accessible N-terminus of the proform of PR3 which folds inwards as a result of the cleavage of the activation dipeptide.
Until specific PR3-ANCA target epitopes are identified, it remains unclear whether the observed different reactivity of patient sera with the proform and the mature form of PR3 is primarily a consequence of shape change of certain epitopes (same amino acid residues), or whether the shape change of the PR3 molecule renders ‘new’ antigenic sites (different amino acid residues) accessible to the ANCA. Consequently, the different reactivity with the two rPR3 substrates reflects either binding to different epitopes or the fact that some PR3-ANCA epitopes are more affected by the conformational change associated with activation of PR3 than others.
A multitude of in vitro studies have implicated PR3-ANCA in the pathogenesis of small vessel vasculitis associated with WG (reviewed in [1,2]). Preliminary clinical observations, suggesting that patients with biopsy-proven WG of the respiratory tract lacking PR3-ANCA suffer less vasculitic complications, also support such a pathogenic role of PR3-ANCA [21]. While c-ANCA IIF titres correlate well with disease activity in many patients, some may have persistently high c-ANCA titres, despite being in complete clinical remission [22,23]. Our findings indicate that different PR3-ANCA-containing sera may contain antibodies reacting with different epitopes, and frequently a mixture thereof. The presence of a mixture of antibodies may explain why test results obtained with a method that does not allow the distinction between the subtypes do not always correlate with clinical disease activity. Furthermore, it is possible that PR3-ANCA reactive with certain epitopes may be more pathogenic than other PR3-ANCA reactive with other epitopes.
Our findings stress the need for identification of the epitopes recognized by PR3-ANCA from certain patients and the need to understand better the impact of the binding of PR3-ANCA to defined epitopes on various PR3 functions. It would also be of interest to evaluate the differential effects of these PR3-ANCA subtypes on various mechanisms proposed to play a pathogenic role in the development of vasculitis, such as oxygen radical release from neutrophils [24–27], chemokine release from neutrophils and monocytes [28–30], or interference with macromolecular substrate cleavage by PR3 [31,32]. Selective testing for the different PR3-ANCA subtypes may also prove to be more useful to monitor clinical disease activity than testing by standard IIF.
Simple assays allowing the subtyping of PR3-ANCA are required for such investigations. Here we show for the first time that non-haematopoietic cells can be stably transfected with specifically designed constructs coding for rPR3 variants with primary sequence and post-synthetic modifications. As these cells only possess a non-regulated secretory pathway, large amounts of the expressed rPR3 variants accumulate in the media supernatant which in turn can serve as a very convenient direct source of target antigen in a simple capture ELISA. Investigations to determine whether the presence of PR3-ANCA reacting with the epitopes that are only exposed on active PR3 correlate with disease activity or certain specific organ manifestations are currently underway in our laboratory.
Acknowledgments
We thank Dr J. H. Butterfield for the kind gift of the HMC-1 cells, Dr A. H. Limper for helpful discussions, and Ms Kathryn L. Streich for help with the preparation of the manuscript. This study was performed during the tenure of a Clinician-Scientist Award from the American Heart Association to U.S. (AHA94004360), and supported by Grants-in-Aid from the American Heart Association (AHA9696008260) and the Minnesota Affiliate of the AHA (MHA147) (both to U.S.), as well as funds from the Mayo Foundation.
REFERENCES
- 1.Kallenberg CGM, Brouwer E, Weening JJ, et al. Anti-neutrophil cytoplasmic antibodies: current diagnostic and pathophysiological potential. Kidney Int. 1994;46:1–5. doi: 10.1038/ki.1994.239. [DOI] [PubMed] [Google Scholar]
- 2.Gross WL, Csernok E. Immunodiagnostic and pathophysiologic aspects of antineutrophil cytoplasmic antibodies in vasculitis. Curr Opin Rheumatol. 1995;7:11–19. [PubMed] [Google Scholar]
- 3.Lüdemann J, Csernok E, Ulmer M, et al. Anti-neutrophil cytoplasm antibodies in Wegener's granulomatosis: immunodiagnostic value, monoclonal antibodies and characterization of the target antigen. Neth J Med. 1990;36:157–62. [PubMed] [Google Scholar]
- 4.Jenne DE, Tschopp J, Lüdemann J, et al. Wegener's autoantigen decoded. Nature. 1990;346:520. doi: 10.1038/346520a0. [DOI] [PubMed] [Google Scholar]
- 5.Gupta SK, Niles JL, McCluskey RT, et al. Identity of Wegener's autoantigen (P29) with proteinase 3 and myeloblastin. Blood. 1990;76:2162. [PubMed] [Google Scholar]
- 6.Sommarin Y, Rasmussen N, Wieslander J. Characterization of monoclonal antibodies to proteinase 3 and application in the study of epitopes for classical anti-neutrophil cytoplasm antibodies. Exp Nephrol. 1995;3:249–56. [PubMed] [Google Scholar]
- 7.Bini P, Gabay JE, Teitel A, et al. Antineutrophil cytoplasmic autoantibodies in Wegener's granulomatosis recognize conformational epitopes on proteinase 3. J Immunol. 1992;149:1409–15. [PubMed] [Google Scholar]
- 8.Szymkowiak CH, Johnston TW, Csernok E, et al. Expression of the human autoantigen of Wegener's granulomatosis (PR3) in a baculovirus expression system. Biochem Biophys Res Commun. 1996;219:283–9. doi: 10.1006/bbrc.1996.0224. [DOI] [PubMed] [Google Scholar]
- 9.Witko-Sarsat V, Halbwachs-Mecarelli L, Almeida RP, et al. Characterization of a recombinant proteinase 3, the autoantigen in Wegener's granulomatosis and its reactivity with anti-neutrophil cytoplasmic autoantibodies. FEBS Letters. 1996;382:130–6. doi: 10.1016/0014-5793(96)00152-4. [DOI] [PubMed] [Google Scholar]
- 10.Fujinaga M, Chernaia MM, Halenbeck P, et al. The crystal structure of PR3, a neutrophil serine proteinase antigen of Wegener's granulomatosis antibodies. J Mol Biol. 1996;261:267–78. doi: 10.1006/jmbi.1996.0458. [DOI] [PubMed] [Google Scholar]
- 11.Specks U, Fass DN, Fautsch MP, et al. Recombinant human proteinase 3, the Wegener's autoantigen, expressed in HMC-1 cells is enzymatically active and recognized by c-ANCA. FEBS Letters. 1996;390:265–70. doi: 10.1016/0014-5793(96)00669-2. [DOI] [PubMed] [Google Scholar]
- 12.Harmsen MC, Heeringa P, Van Der Geld YM, et al. Recombinant proteinase 3 (Wegener's antigen) expressed in Pichia pastoris is functionally active and is recognized by patient sera. Clin Exp Immunol. 1997;110:257–64. doi: 10.1111/j.1365-2249.1997.tb08325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Specks U, Wiegert EM, Homburger HA. Human mast cells expressing recombinant proteinase 3 (PR3) as substrate for clinical testing for anti-neutrophil cytoplasmic antibodies (ANCA) Clin Exp Immunol. 1997;109:286–95. doi: 10.1046/j.1365-2249.1997.4561353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun J, Fass DN, Hudson JA, et al. Capture-ELISA based on recombinant proteinase 3 (PR3) is sensitive for PR3-ANCA testing and allows detection of PR3 and PR3-ANCA/PR3 immunecomplexes. J Immunol Methods. 1998;211:111–23. doi: 10.1016/s0022-1759(97)00203-2. [DOI] [PubMed] [Google Scholar]
- 15.Specks U, Mayer U, Nischt R, et al. Structure of recombinant N-terminal globule of collagen type VI α3 chain and its binding to heparin and hyaluronan. EMBO J. 1992;11:4281–90. doi: 10.1002/j.1460-2075.1992.tb05527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campanelli D, Melchior M, Fu Y, et al. Cloning of cDNA for proteinase 3: a serine protease, antibiotic, and autoantigen from human neutrophils. J Exp Med. 1990;172:1709–15. doi: 10.1084/jem.172.6.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horton RM, Hunt HD, Ho SN, et al. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 18.Williams RC Jr, Staud R, Malone CC, et al. Epitopes on proteinase-3 recognized by antibodies from patients with Wegener's granulomatosis. J Immunol. 1994;152:4722–37. [PubMed] [Google Scholar]
- 19.Chang L, Binos S, Savige J. Epitope mapping of anti-proteinase 3 and anti-myeloperoxidase antibodies. Clin Exp Immunol. 1995;102:112–9. doi: 10.1111/j.1365-2249.1995.tb06644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baslund B, Segelmark M, Wiik A, et al. Screening for anti-neutrophil cytoplasmic antibodies (ANCA): is indirect immunofluorescence the method of choice? Clin Exp Immunol. 1995;99:486–92. doi: 10.1111/j.1365-2249.1995.tb05577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiba M, Specks U. Prognosis of Wegener's granulomatosis limited to the respiratory tract: significance of c-ANCA. Chest. 1996;110:25S. (Abstr.): [Google Scholar]
- 22.Nölle B, Specks U, Lüdemann J, et al. Anticytoplasmic autoantibodies: their immunodiagnostic value. Ann Int Med. 1989;111:28–40. doi: 10.7326/0003-4819-111-1-28. [DOI] [PubMed] [Google Scholar]
- 23.Kerr GS, Fleisher TA, Hallahan CW, et al. Limited prognostic value of changes in antineutrophil cytoplasmic antibody titer in patients with Wegener's granulomatosis. Arthritis Rheum. 1993;36:365–71. doi: 10.1002/art.1780360312. [DOI] [PubMed] [Google Scholar]
- 24.Falk RJ, Terrell RS, Charles LA, et al. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porges AJ, Redecha PB, Kimberly WT, et al. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via FcγRIIa. J Immunol. 1994;153:1271–80. [PubMed] [Google Scholar]
- 26.Brouwer E, Huitema MG, Mulder AHL, et al. Neutrophil activation in vitro and in vivo in Wegener's granulomatosis. Kidney Int. 1994;45:1120–31. doi: 10.1038/ki.1994.149. [DOI] [PubMed] [Google Scholar]
- 27.Reumaux D, Vossebeld PJM, Roos D, et al. Effect of tumor necrosis factor-induced integrin activation on Fcγ receptor II-mediated signal transduction: relevance for activation of neutrophils by anti-proteinase 3 or anti-myeloperoxidase antibodies. Blood. 1995;86:3189–95. [PubMed] [Google Scholar]
- 28.Casselman BL, Kilgore KS, Miller BF, et al. Antibodies to neutrophil cytoplasmic antigens induce monocyte chemoattractant protein-1 secretion from human monocytes. J Lab Clin Med. 1995;126:495–502. [PubMed] [Google Scholar]
- 29.Brooks CJ, King WJ, Radford DJ, et al. IL-1β production by human polymorphonuclear leucocytes stimulated by anti-neutrophil cytoplasmic autoantibodies: relevance to systemic vasculitis. Clin Exp Immunol. 1996;106:273–9. doi: 10.1046/j.1365-2249.1996.d01-835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ralston DR, Marsh CB, Lowe MP, et al. Antineutrophil cytoplasmic antibodies induce monocyte IL-8 release. Role of surface proteinase-3, α1-antitrypsin, and Fcγ receptors. J Clin Invest. 1997;100:1416–24. doi: 10.1172/JCI119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leid RW, Ballieux Bepb, van der Heijden I, et al. Cleavage and inactivation of human C1 inhibitor by the human leukocyte proteinase, proteinase 3. Eur J Immunol. 1993;23:2939–44. doi: 10.1002/eji.1830231132. [DOI] [PubMed] [Google Scholar]
- 32.Robache-Gallea S, Morand V, Bruneau JM, et al. In vitro processing of human tumor necrosis factor-α. J Biol Chem. 1995;270:23688–92. doi: 10.1074/jbc.270.40.23688. [DOI] [PubMed] [Google Scholar]