

Abstract
Human deficiencies of terminal complement components are known to be associated with increased susceptibility to Neisseria meningitidis infection. Polymorphic DNA marker studies in complement deficient investigations allow identification of haplotypes associated with the deficiency and enable the possible identification of heterozygote carriers of the defect. We report studies of an Irish family in which the index case had suffered recurrent meningococcal disease and was found to be deficient in the seventh component of complement (C7). The availability of all family members enabled us to determine the segregating haplotypes. The defects in the family segregated with two very closely related C6 and C7 DNA haplotypes, one of which is known to be associated with the large Irish C7 DNA deletion defect. The index case and two C7 deficient siblings were found to be homozygous for this defect, a deletion that spans approx. 6.8 kbp and encompasses exons 7 and 8. The deletion defect of exons 7 and 8 of C7 has been found in homozygous form in another C7 deficient Irish individual, and is present in heterozygous form in C7 deficient members of a third Irish family. Therefore, this deletion defect occurs in five of the six deficient chromosomes of these three unrelated Irish families, raising the interesting question of how prevalent this defect may be within the Irish community.
Keywords: complement, genetic defects, DNA markers, C7 deficiency, Neisseria meningitidis
INTRODUCTION
The terminal complement system consists of components five to nine. Of these, components six to nine are related plasma proteins which differ in size and complexity [1]. Activation of the complement cascade results in the terminal complement components combining to form the C5b-9 complex. On cell membranes, this complex becomes the membrane attack complex (MAC) which is capable of forming trans-membrane channels through which ions migrate, leading to cell lysis and cell death [2].
Genetically determined human deficiencies of any of the terminal complement components are associated with increased susceptibility to Neisseria meningitidis infections [3–5]. These deficiencies are determined traditionally using haemolytic complement assays [6]. The genes for the sixth and seventh complement components (C6, C7) have been assigned to chromosome 5p13 [7]. These genes have been shown to be linked closely on this chromosome [8,9]. Both the C6 and C7 genes are polymorphic and are close homologues of the C8α, C8β and C9 genes [10]. Molecular bases of C6 and C7 deficiencies have been reported [11–13], as well as those of subtotal deficiencies of C6 and C7 [14].
A rapidly increasing number of polymorphic DNA markers for the C6 and C7 genes have been described [15–19]. DNA marker studies in complement deficient subjects allow the determination of genetic markers associated with deficiencies in individuals who lack protein markers [15,19–21]. Neutral DNA polymorphic markers arise from changes in the DNA sequence, usually by only one base pair, that do not result in the arrest or major alteration of protein synthesis. These are commonly detected by some form of restriction fragment length polymorphism (RFLP) assay. The use of DNA genetic markers can be very informative; neutral genetic markers can be used to detect linked genetic defects, as they directly assess the genotype rather than examine phenotypic features. Consequently, DNA marker studies are useful in detecting heterozygote carriers, as well as being indicators of the genetic heterogeneity of the defects [19,21]. Ten different polymorphic DNA markers have been reported for the C6 and C7 genetic region. These include three Taq I polymorphic sites [15,17,22], a C6 Msp I site [16], four C7 DNA polymorphic sites [17,18,20], a C6 A/B polymorphism [23,24] and a C7 intron 1 Apo I site (Fernie et al. unpublished data). These 10 polymorphic DNA markers potentially generate 1024 different haplotype combinations.
The investigations reported here focused on the study of polymorphic DNA markers in an Irish C7 index case and his immediate family. The deficiency had been ascertained due to repeated meningococcal infections. The availability of all family members in the study enabled us to ascribe a particular DNA marker haplotype to the defective condition. The determination of C6/C7 DNA haplotypes of the index case in the marker study allowed us to proceed immediately to testing and confirming the occurrence of the C7 exon 7 and 8 deletion defect [13] in both his C7 genes. Confirmation that the C7 deletion occurs in a homozygous form in this family now means that this defect has been found in five of six deficient chromosomes in three unrelated families in the Republic of Ireland, posing the possibility that it may be a relatively common cause of C7 deficiency in this country.
SUBJECTS AND METHODS
Subjects
Family A was an Irish family that has not previously been reported and comprised both parents, four sons and four daughters (Fig. 1). The C7 deficient index case, subject II.7, had been ascertained because of meningococcal infections at the age of 16 years and again a year later.
Fig. 1.
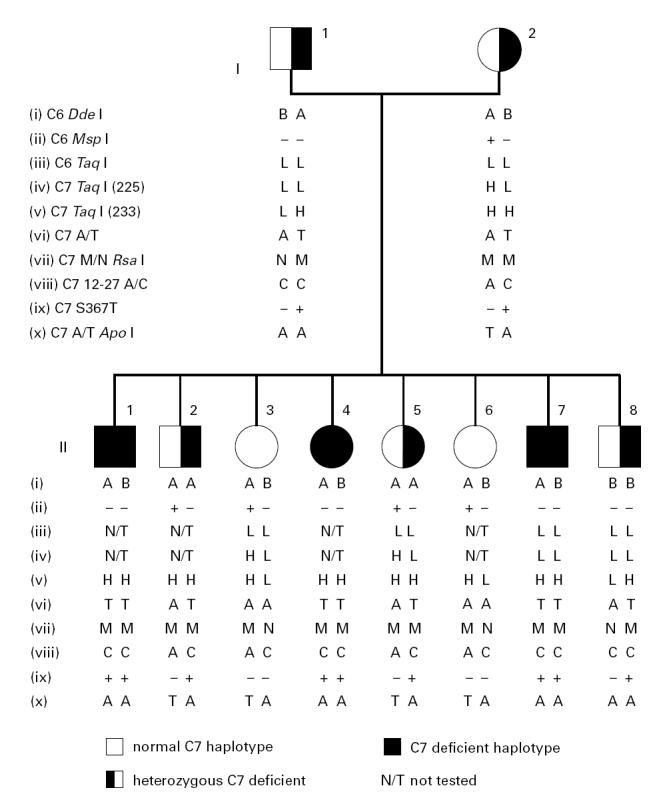
Family A comprised parents and eight children. Subjects II.1, II.4 and II.7 are C7 deficient. The data on the markers are presented as vertical haplotypes.
Functional assays
Total functional complement activity and C7 functional activity in the serum of members of family A were tested by haemolytic assays and double diffusion haemolytic assays in 1% agarose gels [6,25].
DNA preparation
Genomic DNA was isolated for all 10 family members and controls from whole EDTA-treated blood by a modification of a method described previously [26].
Polymerase chain reaction
Exon-specific polymerase chain reaction (PCR) amplification [27] was performed on intron and exon 3 of C6, and on exons 1, 2, 6, 7, 8, 9, 11, 13, 15, 16 and 17 of the 18 C7 exons. Details of the primers used and the annealing temperatures are given in Table 1. Cycle conditions were 93°C for 1 min for denaturation, followed by 1 min at the specified annealing temperature. Extension was performed at 72°C for 1 min over 30 cycles in a Perkin-Elmer Cetus Thermal Cycler (Foster City, CA) or for 2 min at 72°C in an MJ Mini-Cycler (MJ Research Inc., Waterton, MA). dNTPs (50 μmol/l), 0.5 μmol/l of each primer and 0.5 U Taq DNA polymerase (Promega Corp., Madison, WI) were used with about 100 ng of template DNA in a final reaction volume of 50 μl. All amplifications were performed under mineral oil (Sigma-Aldrich Chemical Co., Poole, UK).
Table 1.
Oligonucleotide primers and products of exon polymerase chain reaction (PCR)

Amplification by PCR was confirmed by examination of 5 μl of PCR product on 2% agarose mini-gels in standard 1 × Tris-borate-EDTA buffer (0.09 m Tris-borate, 0.002 m EDTA) (TBE) with 0.5 μg/l ethidium bromide. The gels were subjected to electrophoresis at a constant current of 65 mA and subsequently visualized by ultra violet (UV) illumination.
Restriction digests
Genetic markers in PCR products were examined by restriction digests. The restriction enzymes Dde I, Apo I, Rsa I and Bsr GI were obtained from New England Biolabs (St Albans, UK). Digestion was carried out over 2–4 h at the specified incubation temperature using the supplied buffers according to the manufacturer's instructions.
Digests of C7 exon 9 with Dde I and C7 exon 13 with Bsr GI were examined following electrophoresis on 2% agarose/TBE mini-gels. Smaller DNA fragments were resolved by electrophoresis on either 10% w/v or 12% w/v (19:1 acrylamide:bis-acrylamide ratio) polyacrylamide gels in TBE, cast on Gel-Bond PAG Film (FMC Bioproducts, Rockland, ME). Dde I digested C6 exon 3 and Apo I digested C7 exon 1 PCR products were examined on 12% w/v polyacrylamide gels, whereas Rsa I digested C7 exon 13 and Msp I digested intron 3 of C6 products were resolved on 10% w/v polyacrylamide gels. Following electrophoresis all polyacrylamide gels were silver-stained using an adapted protocol [28] as outlined by Fernie et al. [24].
Southern blotting
Further restriction digests using the enzymes Sac I, Hind III and Taq I (New England Biolabs) were performed on approx. 10 μg of genomic DNA from selected members of family A and normal controls, adhering to the manufacturer's protocol, at a specified incubation temperature and using the buffers supplied. The samples were analysed on 0.8% agarose submarine gels in 0.04 m Tris-acetate/0.001 m EDTA buffer and blotted overnight [29] onto Hybond N filters (Amersham International, Aylesbury, UK). The filters were UV cross-linked in a Stratalinker (Stratagene, Cambridge, UK) before hybridization to 32P-labelled C6 or C7 cDNA probes. The probes were radiolabelled using the Ready-to-Go DNA Labelling Kit (Pharmacia Biotech, St Albans, UK) according to the manufacturer's instructions. Hybridization was conducted under standard conditions [30]. Washing stringency was 0.4 × sodium chloride sodium citrate/0.1% SDS at 65°C and the filters were autoradiographed under enhancing conditions [31].
Investigation of C6/C7 marker haplotypes
DNA marker studies were performed to determine the C6/C7 haplotypes of the family in order to establish if they were likely to be homozygous or heterozygous for the molecular defect. The arrangement of the genes and the positions of the polymorphic markers on the chromosome are shown in Fig. 2.
Fig. 2.

Map of the C6 and C7 gene region of chromosome 5p13 showing the positions of the 10 polymorphic DNA markers used in this study.
The C6 A/B marker was determined by digestion of C6 exon 3 product with Dde I, as the responsible polymorphic site is at position 56 of the exon [23,24]. The PCR product of intron 3 of C6 was digested with Msp I [16]. The markers C6βTaq I (adjacent to C6 exon 17); C7-225 Taq I (C7 intron 15); and C7-233 Taq I (C7 intron 13) (where β, -225 and -233 indicate the C6 or C7 cDNA probe used) were determined via Southern blotting [29] as described earlier [15,17,22]. The C to G polymorphism at position 367 of C7 exon 9 leads to a threonine/serine substitution. The polymorphism was investigated using Dde I digested PCR product (C7 S367T) [17]. The C7 A/T polymorphic marker was determined by digestion of amplified exon 1 and part of intron 1 of C7 with Apo I, as within the amplicon there is a T to A polymorphism at position 289 of intron 1 (Fernie et al., unpublished data).
The PCR product of C7 exon 13 was digested with both Rsa I and Bsr GI. The base change responsible for the C7 M/N protein polymorphism [32], at position 363 of the exon, was detected by Rsa I digestion [33]. The adjacent A → T point substitution at position 396, resulting in the C7 T576S polymorphism, was detected by Bsr GI digestion of the same PCR product [19]. The final marker was a C → A point substitution at position 327 in intron 12 of C7 (C7 12.-27) only 36 bp 5′ of C7 M/N. This marker was detected by the amplification refractory mutation system (ARMS) using allele-specific primers [18,34]. The nomenclature for the alleles investigated in the DNA marker study are those used previously [19]. These 10 bi-allelic markers define 1024 possible haplotypes in the C6/C7 gene region.
RESULTS
Functional assays
Total complement functional assays, C7 functional assays and double diffusion haemolytic assays indicated a lack of functional C7 in subjects II.1, II.4 and II.7 of the family (Fig. 1), whereas all other family members were found to be complement sufficient.
Marker studies
A total of 10 polymorphic DNA markers in the C6 and C7 gene regions were investigated (Fig. 2). The inclusion of all 10 family members in the marker studies enabled us to ascribe a particular DNA marker haplotype to the defective condition and to construct a family tree for family A (Fig. 1). It was found that of the eight siblings; two sons and one daughter are C7 deficient (subjects II.1, II.4 and II.7), two sons and one daughter are heterozygous carriers of the deficiency (subjects II.2, II.5 and II.8), whereas the other two daughters are homozygous C7 sufficient (subjects II.3 and II.6). The proband (subject II.7) and his deficient brother and sister (subjects II.1 and II.4) are homozygous for all C7 markers and all but one of the C6 markers (C6 exon 3, Dde I).
PCR amplifications
The presence of intron and exon 3 of C6, and exons 1, 2, 6, 9, 11, 13, 15, 16 and 17 of C7 was confirmed in all family members by examination of PCR products following electrophoresis. Attempts at PCR amplification of exon 7 and exon 8 of C7 failed in all three C7 deficient siblings, but not in any complement sufficient family members. In order to confirm that the PCR failures of exons 7 and 8 were not due to localized absence or modification of the intron sequences of the template, further PCR reactions were carried out between primers within the exons and those in the flanking introns. These also failed, indicating the probable deletion of both exon 7 and exon 8 in deficient members of family A.
Similar PCR failures of exons 7 and 8 of C7 have been reported for another C7 deficient Irish individual [13], who is homozygous for all of the C6 and C7 markers. The ‘deficient’ haplotype of this subject (Table 2, subject II.2, family 2) is identical to one of those presented here for the index case of family A (Table 2). Therefore, to establish if the molecular basis of the defect in these two families is also identical, we proceeded to analyse for the C7 exon 7 and 8 deletion defect.
Table 2.
C7 deficient haplotypes of three unrelated C7 deficient Irish subjects

Southern blotting—verification of exon deletions
Southern blots using Hind III, Sac I or Taq I restriction digests were used to investigate the extent of the deletion of exons 7 and 8 in the proband. Hind III blots are informative, as exons 8 and 9 are contained in individual Hind III fragments, and in the normal chromosome a Hind III fragment containing exons 6 and 7 is about 14 kbp [13]. The size of this fragment is not significantly different if exons 7 and 8 are deleted, as the fragment then presumably terminates at a Hind III site just 5′ of exon 9 [13]. Probing of a Hind III blot with an exon 7 probe revealed no band in the proband, but hybridized to the normal 14-kbp fragment in complement sufficient family members (data not shown). This confirms a deletion of exon 7 in subject II.7. A similar approach using an exon 8 probe is not practical, as the probe contains repetitive sequences and would hybridize to a smear.
A Taq I blot probed with C7-401 3′ cDNA, which contains exons 4–12, reveals a number of bands including a 1.9-kbp fragment, which is seen in the index case, his parents and heterozygous siblings, but not in subject II.3, a normal sister. Another 2.65-kbp fragment containing the 5′ half of exon 7 is absent in subject II.7, but present in all normal and heterozygous family members examined (data not shown).
The Sac I digest was particularly informative. Subject II.7 of family A produces a band of 12.7 kbp but not the 11.4-kbp band which is revealed by normal individuals (Fig. 3). However, his parents and heterozygote siblings reveal both the 12.7-kbp and 11.4-kbp band fragments (Fig. 3). As reasoned by Fernie et al. [13], a detailed examination of these band fragments and the positions of the restriction sites of Sac I on the exon [13] indicated that the deletion of exons 7 and 8 of C7 spans approx. 6.8 kbp.
Fig. 3.
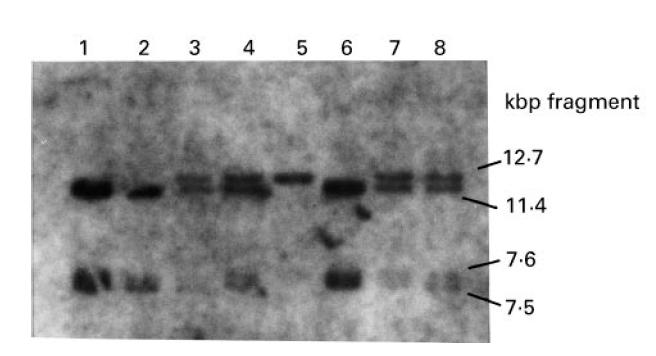
Sac I Southern blot showing the presence of a 12.7-kbp fragment in the proband (lane 5) and a 11.4-kbp fragment in normal individuals (lanes 1, 2, 6). The parents (lanes 3, 4) and heterozygous siblings II.5 and II.8 (lanes 7, 8) reveal both the 11.4-kbp and 12.7-kbp fragments. The 7.5-kbp and 7.2-kbp fragments are revealed due to the presence of other Sac I restriction sites along the chromosome [13].
DISCUSSION
It is possible to provide a complete explanation for the C7 deficiency in family A from the presented data. The apparent deletion of exon 7 and exon 8 of C7, highlighted initially by PCR failure, was subsequently confirmed in two ways. First, PCR reactions between primers within the exons and those in the flanking introns also failed, and second, probing of a Hind III blot with an exon 7 probe revealed no band in affected family members. This is satisfactory confirmation of the absence of a gene segment in these subjects. These results, in addition to those obtained in the Sac I and Taq I digested blots, are similar to those previously published for an unrelated C7 deficient Irish individual (subject II.2, family 2) [13]. Fernie et al. [13] conclude that this deletion of exons 7 and 8 is approx. 6.8 kbp, where the 5′ boundary is 800–1400 bp 5′ of exon 7 and the 3′ boundary is 1200–1800 bp 5′ of exon 9. Such a deletion is sufficiently large to be responsible for the homozygous C7 deficiency in affected members of family A and is the largest defect reported to date in a terminal complement component deficient gene.
The deficient haplotypes of family A are shown in Table 2. Also shown for comparison are the deficient haplotypes of two other unrelated C7 deficient Irish individuals who had suffered recurrent meningococcal infections. These subjects have been reported previously by Fernie et al. [13], as the index cases in families 1 and 2 and their deficient haplotypes had been determined and assigned by similar marker studies [19]. However, the deficient haplotypes of these individuals have now been extended to include the C7 intron 1 Apo I polymorphic site (Table 2).
The C7 deficient members of family A are homozygous for all C7 markers and all but one of the C6 markers (C6 exon 3, Dde I). The deficient haplotype B-L; LLHTMC+A, found in the mother (subject I.2) and four siblings (subjects II.1, II.4, II.7 and II.8) is identical to those reported for subject II.2, family 2 [13]. This subject is homozygous for all of the C6 and C7 markers, including the C7 intron 1 Apo I polymorphic marker not investigated in the previous study. This C7 deficient haplotype is present also in heterozygous form in subject II.1 of family 1 (Table 2) [13].
The other very closely related deficient haplotype of family A, A-L; LLHTMC+A, is present in the father (subject I.1) and five of his offspring (subjects II.1, II.2, II.4, II.5 and II.7). This deficient haplotype differs from the other deficient haplotype of the family and those previously described (subject II.2, family 2 and subject II.1, family 1) [13] in only one polymorphic marker site, C6 exon 3 Dde I A/B. However, given the position of this marker site on the chromosome (Fig. 2), the possibility of a recombination event occurring between this and the next marker site is entirely feasible. Therefore, these two very closely related haplotypes occur in five of the six deficient chromosomes in three unrelated Irish families, and in each instance are associated with the exon 7 and 8 deletion defect. This raises the interesting question of how prevalent the deletion defect may be within the Irish community. The occurrence of the same C7 defect on two different C6/C7 haplotypes does suggest that this is an ancient defect, reaching far back into Irish history [35]. However, we have insufficient data to estimate how ancient the defect is or how long it has been segregating in the Irish population.
C7 deficiency, like C8 deficiency, appears to bear no selective advantage or disadvantage in heterozygotes [36]. Other reported investigations of patients with C7 deficiency reveal a wide variety of C7 haplotypes in sporadic cases of widespread geographical origin. Heterogeneous molecular defects leading to C7 deficiency in Japan have been described, although the two patients were homozygous for their particular defect [12]. However, many of the defects appear homogeneous for individuals living in defined geographical areas [19], and we have observed several predominantly homogeneous C6 and C7 defects among the Moroccan Israelis [19], and in the Western Cape, South Africa [37].
In family A, subjects II.1 and II.4, both C7 deficient, have not to date developed meningococcal infection, highlighting that deficiency is not incompatible with apparently normal health [5]. Diagnosis of underlying deficiency in patients presenting with meningococcal infections does allow appropriate precautions to be taken to minimize the risk of further infections [25]. DNA polymorphisms can be detected irrespective of whether the sequence change affects the phenotype, and prior knowledge of the molecular defect responsible for the deficiency is not required. Thus, DNA marker studies in investigations of affected families enables identification of both heterozygous and homozygous individuals. Identification of heterozygous carriers within affected families allows for the provision of informed genetic counselling and medical advice. Investigations of complement function and identification of possible complement deficiencies are important as part of the immunological assessment of subjects showing increased susceptibility to serious infections, including infections by N. meningitidis.
Acknowledgments
This study was supported by grants from the HRB/British Council Programme 1997 under the Irish/UK Research Visits Scheme (to A.M.O'H.), the Irish Health Research Board (to A.O. and A.P.M.), and the Meningitis Research Foundation (to A.O.).
References
- 1.Müller-Eberhard HJ. The membrane attack complex of complement. Annu Rev Immunol. 1986;4:503–28. doi: 10.1146/annurev.iy.04.040186.002443. [DOI] [PubMed] [Google Scholar]
- 2.Esser AF. C9-mediated cytotoxicity and the function of poly (C9) In: Bonavida B, Collier RJ, editors. Membrane-mediated cytotoxicity. UCLA Symposia on Molecular and Cellular Biology. Vol. 45. New York: New Series, Liss; 1987. p. 411. [Google Scholar]
- 3.Ross SC, Denson P. Complement deficiency status: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine. 1984;63:243–73. [PubMed] [Google Scholar]
- 4.Potter PC, Frasch CE, van der Sande WJM, Cooper RC, Patel Y, Orren A. Prophylaxis against Neisseria meningitidis infections and antibody responses in patients with deficiency of the sixth component of complement. J Infect Dis. 1990;161:932–7. doi: 10.1093/infdis/161.5.932. [DOI] [PubMed] [Google Scholar]
- 5.Würzner R, Orren A, Lachmann PJ. Inherited deficiencies of the terminal components of human complement. Immunodefic Rev. 1992;3:123–47. [PubMed] [Google Scholar]
- 6.Harrison A, Lachmann PJ. Complement technology. In: Weir DM, editor. Handbook of experimental immunology. 4. Vol. 1. Oxford: Blackwell Scientific Publications; 1986. pp. 39.1–39.49. [Google Scholar]
- 7.Jeremiah SJ, Abbot CM, Murad Z, Povey S, Thomas HJ, Solomon E, DiScipio R, Fey GH. The assignment of the genes coding human complement components C6 and C7 to chromosome 5. Ann Hum Genet. 1990;54:141–7. doi: 10.1111/j.1469-1809.1990.tb00370.x. [DOI] [PubMed] [Google Scholar]
- 8.Hobart MJ, Fernie BA, DiScipio RG, Lachmann PJ. A physical map of the C6 and C7 complement component gene region on chromosome 5p13. Hum Molec Genet. 1993;2:1035–6. doi: 10.1093/hmg/2.7.1035. [DOI] [PubMed] [Google Scholar]
- 9.Setien F, Alvarez V, Coto E, DiScipio RG, Lopez-Larrea C. A physical map of the human complement component C6, C7 and C9 genes. Immunogenetics. 1993;38:341–4. doi: 10.1007/BF00210475. [DOI] [PubMed] [Google Scholar]
- 10.Hobart MJ, Fernie BA, DiScipio RG. Structure of the human C7 gene and comparison with the C6, C8A, C8B and C9 genes. J Immunol. 1995;154:5188–94. [PubMed] [Google Scholar]
- 11.Nishizaka H, Horiuchi T, Zhu Z-B, et al. Molecular bases of inherited human complement component C6 deficiency in two unrelated individuals. J Immunol. 1996;156:2309–15. [PubMed] [Google Scholar]
- 12.Nishizaka H, Horiuchi T, Zhu Z-B, Fukumori Y, Volonakis JE. Genetic bases of human complement C7 deficiency. J Immunol. 1996;157:4239–43. [PubMed] [Google Scholar]
- 13.Fernie BA, Orren A, Sheehan G, Schlesinger M, Hobart MJ. Molecular bases of C7 deficiency: three different defects. J Immunol. 1997;159:1019–26. [PubMed] [Google Scholar]
- 14.Fernie BA, Würzner R, Orren A, et al. Molecular bases of combined subtotal deficiencies of C6 and C7 and their effects in combination with other deficiencies. J Immunol. 1996;157:3648–57. [PubMed] [Google Scholar]
- 15.Coto E, Martinez-Navez E, Dominguez O, DiScipio R, Urra JM, Lopez-Larrea C. DNA polymorphisms and linkage relationship of the human complement C6, C7 and C9 genes. Immunogenetics. 1991;33:184–7. doi: 10.1007/BF01719238. [DOI] [PubMed] [Google Scholar]
- 16.Fernie BA, Finlay A, Price D, Chan E, Orren A, Joysey VC, Joysey KA, Hobart MJ. Complement C6 and C7 DNA polymorphisms analysed by PCR in seven ethnic groups and characterisation of the C6 Msp I RFLP. Exp Clin Immunogenet. 1996;13:92–103. [PubMed] [Google Scholar]
- 17.Fernie BA, Würzner R, Unsworth DJ, Tuxworth RI, Hobart MJ. DNA polymorphisms of the complement C6 and C7 genes. Ann Hum Genet. 1995;59:163–81. doi: 10.1111/j.1469-1809.1995.tb00739.x. [DOI] [PubMed] [Google Scholar]
- 18.Fernie BA, Würzner R, Orren A, Hobart MJ. A new intronic polymorphism in the C7 gene 36 bp from the common expressed C7 M/N polymorphism. Ann Hum Genet. 1996;60:179–82. doi: 10.1111/j.1469-1809.1996.tb01187.x. [DOI] [PubMed] [Google Scholar]
- 19.Fernie BA, Orren A, Schlesinger M, Würzner R, Platonov AE, Cooper RC, Williams YE, Hobart MJ. DNA haplotypes of the complement C6 and C7 genes associated with deficiencies of the seventh component; and a new DNA polymorphism in C7 exon 13. Ann Hum Genet. 1997;61:287–98. doi: 10.1046/j.1469-1809.1997.6140287.x. [DOI] [PubMed] [Google Scholar]
- 20.Dewald G, Nöthen MM, Rüther K. A common Ser/Thr polymorphism in the perforin homologous region of human complement component C7. Hum Hered. 1994;44:301–4. doi: 10.1159/000154235. [DOI] [PubMed] [Google Scholar]
- 21.Fernie BA, Orren A, Würzner R, Jones AM, Potter PC, Lachmann PJ, Hobart MJ. Complement component C6 and C7 haplotypes associated with deficiencies of C6. Ann Hum Genet. 1995;59:183–95. doi: 10.1111/j.1469-1809.1995.tb00740.x. [DOI] [PubMed] [Google Scholar]
- 22.Coto E, Martinez-Navez E, Dominguez O, Urra JM, Rodriguez V, Lopez-Larrea C. Taq I polymorphism in the complement component of the C7 gene. Nucl Acids Res. 1990;18:1929. doi: 10.1093/nar/18.7.1929-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dewald G, Nöthen M, Cichon S. Polymorphism of human complement component C6: amino acid substitution (Glu/Ala) within the second thrombospondin repeat differentiates between the two common allotypes C6A and C6B. Biochem Biophys Res Commun. 1993;194:458–64. doi: 10.1006/bbrc.1993.1841. [DOI] [PubMed] [Google Scholar]
- 24.Fernie BA, Delbridge G, Hobart MJ. Correlation of a Glu/Ala substitution at position 98 with the complement C6 A/B phenotypes. Hum Molec Genet. 1993;2:591–2. doi: 10.1093/hmg/2.5.591. [DOI] [PubMed] [Google Scholar]
- 25.Egan L, Orren A, Doherty J, Würzner R, McCarthy CF. Hereditary deficiency of the seventh component of complement and recurrent meningococcal infection: investigations in an Irish family using a novel haemolytic screening assay for complement activity and C7 M/N allotyping. Epidemiol Infect. 1994;113:275–81. doi: 10.1017/s0950268800051700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeffreys AJ. DNA sequence variants in the Gγ-, Aγ-. δ- and β- globin genes in man. Cell. 1979;18:1–10. doi: 10.1016/0092-8674(79)90348-9. [DOI] [PubMed] [Google Scholar]
- 27.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Primer-directed enzymic amplification of DNA with thermostable DNA polymerase. Science. 1988;239:487–91. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 28.Merril CR, Goldman D, Sedman SA, Ebert MH. Ultrasensitive stain for proteins in polyacrylamide gels shows regional variation in cerebrospinal fluid protein. Science. 1981;211:1437–8. doi: 10.1126/science.6162199. [DOI] [PubMed] [Google Scholar]
- 29.Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975;98:503–17. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook K, Fritsch EF, Maniatis T. A laboratory manual. 2. Cold Spring Harbour, NY: Cold Spring Harbour Laboratory Press; 1989. Molecular cloning. [Google Scholar]
- 31.Laskey RA, Mills AD. Enhanced autoradiographic detection of 32P and 125I using intensifying screens and hypersensitized film. FEBS Letters. 1977;82:314–6. doi: 10.1016/0014-5793(77)80609-1. [DOI] [PubMed] [Google Scholar]
- 32.Würzner R, Hobart MJ, Orren A, Tokunaga K, Nitze R, Gotze O, Lachmann PJ. A novel protein polymorphism of human complement C7 detected by a monoclonal antibody. Immunogenetics. 1992;35:398–402. doi: 10.1007/BF00179797. [DOI] [PubMed] [Google Scholar]
- 33.Würzner R, Fernie BA, Jones AM, Lachmann PJ, Hobart MJ. Molecular basis of the complement MN polymorphism—a neutral amino acid substitution outside the epitope of the allospecific monoclonal antibody. J Immunol. 1995;154:4813–9. [PubMed] [Google Scholar]
- 34.Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, Markham AF. Analysis of any point mutation in DNA. The amplification refractory mutations system (ARMS) Nucl Acids Res. 1989;17:2503–16. doi: 10.1093/nar/17.7.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serre JL, Simon-Buoy B, Mornet E, et al. Studies of RFLP closely linked to cystic fibrosis locus throughout Europe lead to new considerations in populations genetics. Hum Genet. 1990;84:449–54. doi: 10.1007/BF00195818. [DOI] [PubMed] [Google Scholar]
- 36.Platonov AE, Shipulin GA, Shipulina OJ, Vershinina IV, Densen P. Heterozygous C8α complement deficiency does not predispose to meningococcal disease. Clin Exp Immunol. 1997;108:497–9. doi: 10.1046/j.1365-2249.1997.3711263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hobart MJ, Fernie BA, Fijen CAP, Orren A. The molecular bases and ancestry of C6 deficiency in the Western Cape, South Africa. Hum Genet. 1998. (accepted for publication) [DOI] [PubMed]