

Abstract
Vascular injury in vasculitis may be due to activation of circulating neutrophils resulting in their increased adhesiveness to locally activated endothelium (Shwartzman phenomenon). Previously, we demonstrated up-regulation of endothelial intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in biopsies from patients with ANCA-associated vasculitis. In the present study, we investigated the expression of adhesion molecules (CD11b, ICAM-1, VLA-4, l-selectin) and activation markers (CD66b, CD64, CD63) on circulating neutrophils from patients with ANCA-associated vasculitis in comparison with their expression on cells from healthy volunteers and patients with sepsis. We related these findings to parameters of disease activity. Surface marker expression was determined by using a non-activating whole blood flow cytometric assay. The expression of activation markers, but not the expression of adhesion molecules, was increased on neutrophils from patients with active vasculitis. The expression of CD63 and CD66b on neutrophils correlated with disease activity as determined by the Birmingham Vasculitis Activity Score (BVAS). In contrast to patients with active vasculitis, patients with sepsis showed up-regulation of all markers, including adhesion molecules, suggesting that circulating neutrophils are fully activated in sepsis. We conclude that in ANCA-associated vasculitis, circulating neutrophils are not fully activated, since they do not express increased levels of adhesion molecules as sepsis or in the Shwartzman reaction. These findings are compatible with the concept that in vivo vascular damage in ANCA-associated vasculitides does not occur due to a Shwarzman-like reaction but only after ANCA-induced neutrophil activation at the endothelial cell surface.
Keywords: ANCA, neutrophil activation, vasculitis, sepsis, adhesion molecules, flow cytometry
INTRODUCTION
Primary or idiopathic vasculitic syndromes form a group of inflammatory disorders of presumed autoimmune origin characterized by inflammation and necrosis of blood vessels, frequently in combination with granuloma formation. Within the spectrum of vasculitides, small vessel vasculitides, such as Wegener's granulomatosis, microscopic polyangiitis, and Churg–Strauss syndrome, are strongly associated with ANCA. ANCA in those disorders are generally directed against proteinase 3 (PR3) and myeloperoxidase (MPO) [1,2] and, occasionally, against human leucocyte elastase (HLE) [3].
The pathophysiology of idiopathic small vessel vasculitis is not completely understood. In vitro experiments have shown that ANCA have the potential to activate pre-activated neutrophils to the production of reactive oxygen species and the release of lytic enzymes [4–8]. Furthermore, in vitro, ANCA induce increased expression of CD11b on neutrophils [9].
During an inflammatory response in vivo, adhesion molecules mediate the interaction of neutrophils with endothelial cells. Pre-activated neutrophils roll over the endothelial monolayer, where these cells become further activated by locally presented inflammatory mediators. Subsequently, these cells firmly adhere to and transmigrate through the endothelium [10]. In general, during the process of adhesion and transmigration, endothelial cells are not or only minimally damaged.
In vasculitis, various mechanisms of vascular injury, i.e. endothelial cell damage, may be operative. In secondary vasculitic syndromes activation of neutrophils occurs via the activation of the complement cascade after local deposition of immune complexes. However, in ANCA-associated vasculitides immune complexes can not be demonstrated. One of the possible mechanisms of vascular damage in these syndromes is a Shwartzman-like phenomenon in which activation of neutrophils occurs intravascularly [11,12]. This intravascular activation results in up-regulation of CD11b/CD18 expression [13,14], followed by degranulation and production of oxygen radicals once these cells adhere to locally activated endothelium. Previously it has been suggested that this mechanism is operative in lesional tissue of patients with systemic lupus erythematosus (SLE) that lack immune complex depositions [15]. Indeed, in these patients up-regulation of CD11b/CD18 has been demonstrated on circulating neutrophils [13,16].
In the present study, we tested the hypothesis that a Shwartzmann-like phenomenon might also be operative in ANCA-associated vasculitides by analysing the expression of CD11b, other adhesion molecules, and markers of activation on circulating neutrophils in both patients with active ANCA-associated vasculitis and patients in remission. We compared neutrophil activation in patients with vasculitis with that in patients with sepsis, a condition in which intravascular activation of circulating neutrophils has already been demonstrated [17]. In addition, we correlated the extent of neutrophil activation in patients with vasculitis with parameters of disease activity.
PATIENTS, MATERIALS AND METHODS
Patients and controls
The patient group consisted of consecutive patients admitted to our hospital or seen at the out-patient clinic with a diagnosis of WG, Churg–Strauss syndrome (CSS) or microscopic polyangiitis (MPA). All patients were positive for ANCA. Twenty patients with sepsis served as positive controls for leucocyte activation. All patients with sepsis entered the study within hours after admittance to the intensive care unit. Healthy laboratory personel served as normal controls. This study was approved by the Institutional Review Board.
Diagnostic criteria
The diagnoses of WG, CSS and MPA were established according to clinical and histological criteria [2]. All patients fulfilled the Chapel Hill Consensus Conference definitions for WG, CSS and/or MPA [18]. Patients had either active or inactive disease. Patients with active disease had newly developed disease (N) or relapsing disease (R). Relapsing disease was defined as previously described [19,20]. Criteria for relapsing disease are given in Table 1. Patients with newly developed disease were studied before treatment was started. Patients with relapsing disease were studied before treatment was instituted or intensified. Complete remission was defined as the absence of signs or symptoms attributable to active vasculitic disease. Sepsis was defined as previously described [21].
Table 1.
Criteria for relapsing disease

Disease activity
Vasculitis disease activity was measured according to the Birmingham Vasculitis Activity Scoring index (BVAS) [22], whereas vasculitic damage was measured according to the Vascular Damage Index [23]. Severity of sepsis was scored with the Acute Physiology and Chronic Health Evaluation (APACHE) II scoring system [24].
C-reactive protein (CRP) concentrations were measured by using a particle-enhanced nephelometric method and NA latex CRP reagents (Behring, Marburg, Germany).
ANCA detection
ANCA were detected by indirect immunofluorescence on ethanol-fixed granulocytes as previously described [3] using FITC-labelled goat anti-human IgG (Dako, Glostrup, Denmark) in a 1:50. dilution. Test or control sera were tested at a dilution of 1:20, and further at two-fold dilutions. Slides were read by two independent observers, and a titre ≥ 40 was considered positive.
The specificity of ANCA for either PR3, MPO or HLE was detected by capture ELISA as previously described [25]. Anti-lactoferrin antibodies were detected by ELISA on plates directly coated with lactoferrin (5 μg/ml; Sigma, St Louis, MO) as described [26]. Sera were considered positive for one of the above mentioned specificities when values exceeded the mean + 2 s.d. of normal controls (n = 50).
Surface marker analysis of activation markers by flow cytometry
To avoid in vitro activation of granulocytes we used a whole blood method [27–29]. EDTA anti-coagulated blood was kept on ice until sample preparation. Sample preparation was started always within 5 min after blood sampling. All steps were performed in Hanks' balanced balt solution (HBSS) without calcium and magnesium (Gibco, Life Technologies Ltd, Paisley, UK), supplemented with 1% bovine serum albumin (BSA; Boseral, Organon Teknika, Boxtel, The Netherlands). Cells were fixed with 1% paraformaldehyde in PBS for 10 min on ice, washed, followed by two times erythrocyte lysis with lysis buffer (155 mm NH4Cl, 10 mm KHCO3, 0.1 mm Na2EDTA.H2O) for 5 min at 37°C. A panel of MoAbs to leucocyte surface antigens was used for the analysis of leucocyte activation (Table 2). The first antibody was incubated for 1 h at 4°C. After washing, the cells were incubated with a goat anti-mouse immunoglobulin polyclonal antibody conjugated with PE (Southern Biotechnology Associates Inc., Birmingham, AL), 1:20 diluted, supplemented with 5% normal goat serum and 5% normal human serum, for 30 min at 4°C in the dark. Subsequently, cells were washed and stored until flow cytometric analysis was performed.
Table 2.
Activation markers and adhesion molecules on neutrophils

Analysis of surface marker expression was performed on a Coulter Epics ELITE flow cytometer (Coulter, Hialeah, FL), the same day or occasionally the next day (within 18 h). When the cell pellet contained erythrocytes, the intercalating dye, LDS751 (Exiton Chemical, Dayton, OH) was added before flow cytometry measurement. Erythrocytes could successfully be excluded from the leucocyte population in the LDS751/forward scatter dot plot when combined with a life gate. Neutrophils were identified by forward and sideways scatter. Eosinophils were excluded from the neutrophil population by their high autofluorescence. Data were analysed using Immuno-4 software [30].
QC3 beads (Flow Cytometry Standards, Leiden, The Netherlands) were used to calibrate the flow cytometer [31]. However, batch to batch quality of those beads varied remarkably, and within one batch fluorescence intensity diminished over time. Therefore, we decided to compare the results obtained in patients with the results obtained in healthy age-matched volunteers who were analysed in parallel [29].
The expression of surface markers was calculated as mean fluorescence intensity (MFI), corrected for non-specific binding of an irrelevant antibody and the conjugate (NSB), in combination with a percentage of positive cells (pos percentage). The percentage of positive cells was defined as the percentage of cells of the cell population gated in the forward/sideways scatter dot blot with a higher MFI than the NSB stained population. Data were expressed as a percentage of the value obtained from the healthy control who was tested in parallel, according to the following formula:
 |
In order to assess the variability of the normal population, 10 healthy controls, with the same age distribution as the patient population, were analysed simultaneously. Their individual data were expressed as a percentage of the mean of the healthy control population.
Statistical analysis
Groups were analysed for differences in surface expression by means of the Kruskal–Wallis test. Subsequently, differences between groups were analysed by the Mann–Whitney test. Correlation between parameters was analysed by the Spearman rank correlation test. The paired Wilcoxon test was used to test differences between paired observations. These tests were performed by using GraphPad Instat2 Software. A two-tailed P value < 0.05 was considered to indicate statistical significance.
RESULTS
Patients
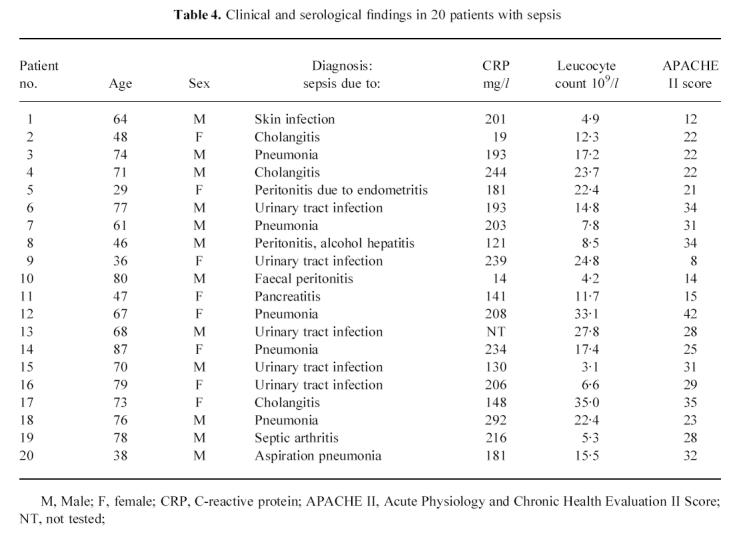
Twenty-eight patients (15 female, 13 male, mean age 53 years, range 22–85 years) with active ANCA-associated vasculitis, i.e. either newly developed disease (n = 14) or relapsing disease (n = 14), were included in this study. At the time of admittance, patients with active disease had a median BVAS score of 13, range 1–39; of these, patients with newly diagnosed disease had a median BVAS score of 20, range 8–39, whereas patients with relapsing disease had a median BVAS score of 9.5, range 1–17 (P < 0.001). Additionally, 12 of these patients were analysed also at times of quiescent disease. Twenty patients with sepsis (12 male, eight female, median age 69 years, range 29–87 years) were included as positive controls for leucocyte activation. At the time of diagnosis, patients had a median APACHE II score of 26, range 8–42. Patient characteristics are given in Table 3 for patients with vasculitis and Table 4 for patients with sepsis
Table 3.
Clinical and serological findings in 28 patients with systemic vasculitis in different stages of disease activity
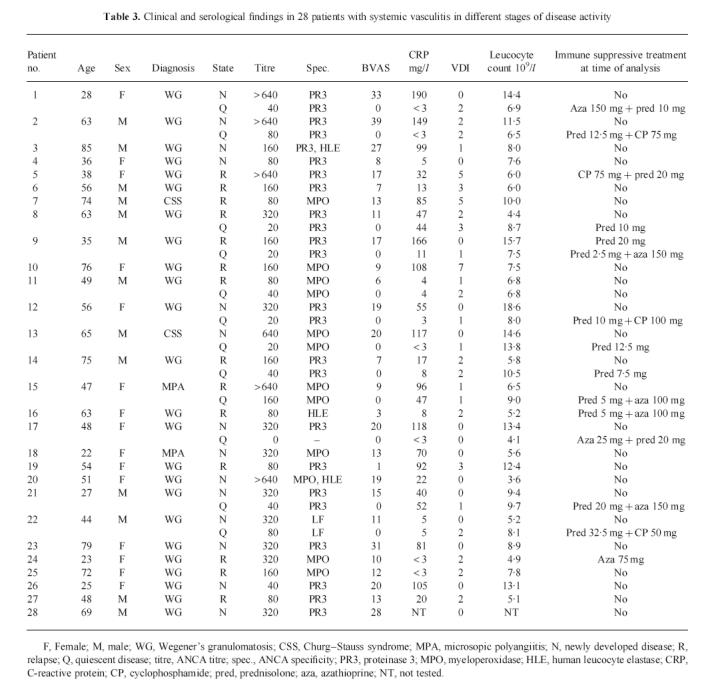
Table 4.
Clinical and serological findings in 20 patients with sepsis
The expression of adhesion molecules on neutrophils
On neutrophils, the expression of CD11b, the α subunit of the Mac-1 integrin, did not differ between the total population of patients with active vasculitis and healthy controls. Expression during active disease did not differ from that during quiescent disease. However, in patients with newly diagnosed vasculitis an increased expression of CD11b (P = 0.04) was found, whereas in patients with relapsing disease the expression of CD11b was not higher than that of healthy controls. CD11b expression on neutrophils from patients with sepsis was increased compared with patients with active vasculitis (P < 0.01) (Fig. 1a).
Fig. 1.
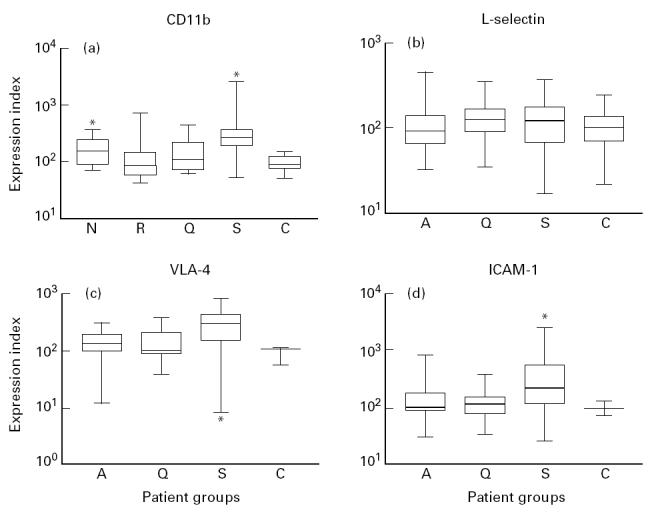
Box and whisker plots indicating the overall range (error bars), 25–75% range (boxes), and median value (horizontal lines) of the surface expression of adhesion molecules (CD11b, VLA-4, intercellular adhesion molecule-1 (ICAM-1) and l-selectin) on neutrophils from patients with active (A), newly diagnosed (N), relapsing (R), and quiescent (Q) ANCA-associated vasculitis, patients with sepsis (S) and healthy controls (C).
*P < 0.05 compared with healthy controls.
In vitro, during cell activation, l-selectin is shed from the cell surface [32,33]. We did not find differences in expression of l-selectin on neutrophils between patients with active disease, sepsis, and healthy controls (Fig. 1b). However, the expression of l-selectin correlated inversely with the BVAS score (r = −0.34, P = 0.04). Moreover, analysis of paired data showed that the l-selectin expression in patients at the time of active disease tended to be decreased compared with its expression during remission (P = 0.09).
VLA-4, a β1-integrin, is expressed mainly on monocytes, lymphocytes and eosinophils [34], but can also be demonstrated on neutrophils [35]. VLA-4 expression on neutrophils did not differ between patients with various stages of disease activity. Neutrophils from patients with sepsis, however, had an increased expression of VLA-4 compared with neutrophils from patients with active vasculitis (P = 0.01) and healthy controls (P = 0.02) (Fig. 1c).
ICAM-1 is expressed on a wide variety of cells, including monocytes and neutrophils [36,37]. ICAM-1 expression on neutrophils did not differ between patients with vasculitis and healthy controls. In patients with sepsis, however, the expression of ICAM-1 was higher than that in patients with active vasculitis (P = 0.02) and healthy controls (P = 0.03) (Fig. 1d). No differences were found in the expression of l-selectin, VLA-4 and ICAM-1 between patients with newly diagnosed disease or relapsing disease and healthy controls. No correlations were found between the extent of expression of CD11b, l-selectin, ICAM-1 and VLA-4 and parameters of disease activity.
Other neutrophil activation markers
The expression of degranulation markers, such as CD66b and CD63, is increased upon activation. CD66b and CD63 are stored in the specific and the azurophilic granules, respectively [38–40]. Patients with active disease showed a higher expression of CD66b on neutrophils than patients with quiescent disease (P = 0.03) and healthy controls (P < 0.01). In patients with sepsis the expression was even higher compared with patients with active vasculitis (P < 0.0001) (Fig. 2a). Analysis of paired data showed that the expression of CD66b during active disease was higher than during remission (P < 0.01) (Fig. 2b). Furthermore, in patients with active vasculitis the expression of CD66b correlated with the BVAS score (r = 0.64 and P < 0.001; Fig. 2c) and CRP values (r = 0.37, P = 0.02), but not with the ANCA titre (P < 0.05).
Fig. 2.

(a) Box and whisker plots indicating the overall range (error bars), 25–75% range (boxes), and median value (horizontal lines) of the surface expression of CD66b on neutrophils from patients with active (A), and quiescent (Q) ANCA-associated vasculitis, patients with sepsis (S) and healthy controls (C). **P < 0.01; ***P < 0.001 compared with healthy controls; †P < 0.05 compared with patients with quiescent disease. (b) Paired analysis of the expression of CD66b on neutrophils from patients with active and quiescent disease. (c) Correlation between the expression of CD66b on neutrophils and disease activity as expressed by the Birmingham Vasculitis Activity Score (BVAS).
Patients with active disease demonstrated a higher expression of CD63 on neutrophils than healthy controls (P < 0.01), but not compared with patients in remission. In patients with sepsis the expression of CD63 was higher than in patients with active vasculitis (P < 0.01) (Fig. 3a). Analysis of paired data showed that the expression of CD63 during active disease was higher than during remission, but this difference did not reach statistical significance (P = 0.11). In patients with active vasculitis the expression of CD63 correlated with the BVAS score (r = 0.40, P = 0.04, Fig. 3b) and CRP values (r = 0.37, P = 0.02), but not with the ANCA titre (P < 0.05).
Fig. 3.
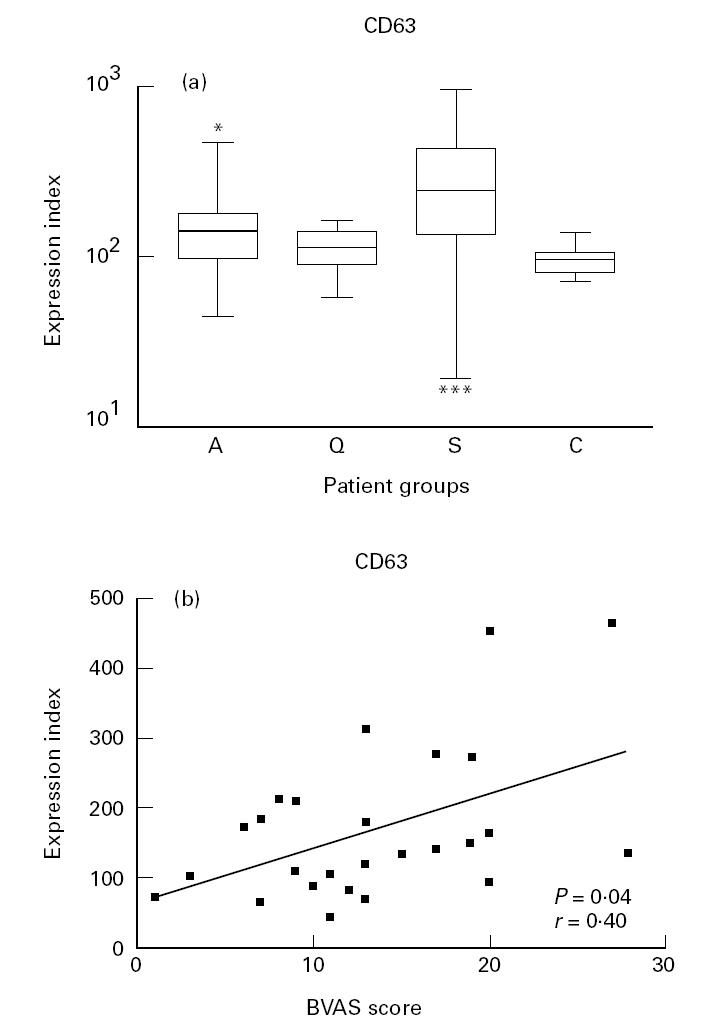
(a) Box and whisker plots indicating the overall range (error bars), 25–75% range (boxes), and median value (horizontal lines) of the surface expression of CD63 on neutrophils from patients with active (A), and quiescent (Q) ANCA-associated vasculitis, patients with sepsis (S) and healthy controls (C). *P < 0.05; ***P < 0.001 compared with healthy controls. (b) Correlation between the expression of CD63 on neutrophils and disease activity as expressed by the Birmingham Vasculitis Activity Score (BVAS).
Expression of CD64, the first Fcγ receptor, is increased upon activation by interferon-gamma (IFN-γ) or granulocyte colony-stimulating factor (G-CSF) [41]. Patients with active disease had an increased expression of CD64 on neutrophils compared with healthy controls (P = 0.0062), but not compared with patients with quiescent disease. In patients with sepsis the expression of CD64 was higher than in patients with active vasculitis (P < 0.0001) (Fig. 4). The expression of CD64 did not correlate with parameters of disease activity or the ANCA titre.
Fig. 4.
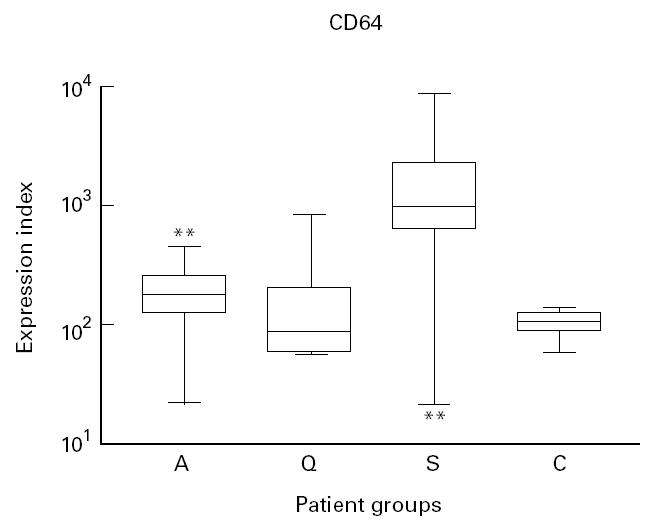
Box and whisker plots indicating the overall range (error bars), 25–75% range (boxes), and median value (horizontal lines) of the surface expression of CD64 on neutrophils from patients with active (A), and quiescent (Q) ANCA-associated vasculitis, patients with sepsis (S) and healthy controls (C).
**P < 0.01 compared with healthy controls.
DISCUSSION
In the present study we tested the hypothesis that a Shwarzman-like reaction underlies vascular damage in patients with ANCA-associated vasculitides. We analysed the expression of different activation markers and adhesion molecules on circulating neutrophils. Furthermore, we compared neutrophil activation in patients with vasculitis with that in sepsis, a condition in which it has been demonstrated that circulating neutrophils are intravascularly activated [17].
In the classical Shwartzman reaction an initial intradermal injection of endotoxin or a comparable stimulant results in local activation of endothelial cells. A subsequent intravenous injection of endotoxin results in complement activation and intravascular neutrophil activation, as demonstrated by the increased expression of CD11/CD18 [14]. Intravascular neutrophil activation eventually leads to endothelial damage as a result of degranulation and production of oxygen radicals once these cells adhere to locally activated endothelium.
In this study, the expression of CD11b and other adhesion molecules on circulating neutrophils was investigated. In inflammation, adhesion molecules play an important role in the interaction between neutrophils and the endothelial monolayer. This interaction is thought to be important for the development of endothelial damage [42]. The expression of CD11b, l-selectin and other adhesion molecules on circulating neutrophils from patients with active vasculitis was not different from their expression in healthy controls, although a slight up-regulation of CD11b was observed in patients with newly diagnosed disease but not in patients with relapsing disease. In contrast, patients with sepsis had significantly increased expression of adhesion molecules on circulating neutrophils, indicating that in these patients cells are activated in the circulation. Several authors have studied the extent of activation of circulating neutrophils in patients with WG. Riecken and co-workers demonstrated a decreased expression of l-selectin compared with healthy controls [43]. These authors, however, used density gradient-isolated granulocytes, which may result in in vitro activation, including shedding of l-selectin, due to purification methods [27,44,45]. In the present study, we used a whole blood method, in which cells were fixed with paraformaldehyde within minutes after collection. All preparation steps were carried out at 4°C. This method minimizes artificial in vitro cell activation, when compared with various cell isolation procedures [28]. Using a non-activating whole blood method, Haller and co-workers also found increased expression of CD11b in patients with newly diagnosed, active vasculitis [46]. In our study we found that CD11b expression was moderately increased only in patients with newly diagnosed active disease, who had more active disease than patients with relapsing disease, as demonstrated by BVAS scores, probably due to delayed diagnosis of progressively active vasculitis. A Shwartzman-like phenomenon may marginally contribute to vascular damage in those patients only who present with ongoing and increasing disease activity. Since we observed that CD11b expression was not increased in relapsing patients, despite the presence of vasculitic lesions, we conclude that other mechanisms of vascular damage are probably more relevant in the pathophysiology of ANCA-associated vasculitides. Previously, Reumeax et al. showed that in vitro ANCA-induced leucocyte activation only occurs when pre-activated cells are bound to a surface, e.g. activated endothelial cells [47]. This mechanism requires pre-activation of circulating neutrophils as well as possibilities for interaction with endothelial cells, e.g. by l-selectin.
In order to investigate whether circulating neutrophils in patients with vasculitis are pre-activated, we assessed the expression of activation markers, such as the degranulation markers CD66b and CD63 and the first Fcγ receptor, CD64, as well as adhesion molecules on those cells. In patients with active vasculitis the expression of CD66b, CD63 and CD64 on neutrophils was increased compared with healthy controls. Patients with active vasculitis lacked, however, increased expression of adhesion molecules on circulating neutrophils. These findings are consistent with a state of pre-activation. Several investigators demonstrated in vitro that pre-activation of neutrophils with cytokines leads to PR3 membrane expression [48,49]. A pre-activated state, i.e. availability of ANCA antigens for interaction with ANCA, is a prerequisite for interaction with and subsequent activation by ANCA. Expression of PR3 can only be explained if a certain degree of degranulation of azurophilic granules has occurred. Indeed, we demonstrated that circulating neutrophils from patients with ANCA-associated vasculitides have an increased expression of CD63, a marker for azurophilic granule degranulation. Furthermore, the expression of these degranulation markers in patients with vasculitis correlated with disease activity, indicating a role for in vivo neutrophil pre-activation in the pathophysiology of ANCA-associated vasculitides.
However, no correlations were found between the extent of activation of circulating neutrophils and ANCA titre. As discussed earlier, we believe that activation of primed neutrophils by ANCA occurs when these cells are bound to a surface, i.e. endothelial cells. Since we investigated the extent of activation of circulating cells, activation of these cells by ANCA had probably not yet occurred. This may explain why no correlation between cell activation and ANCA titre was observed.
The differences in neutrophil activation between patients with active and quiescent disease could, in part, be explained by the effects of treatment of patients with quiescent disease. immunosuppressive drugs are known to influence leucocyte activation. Since it is the treatment that induces quiescent disease and since we believe that it is the activated leucocyte that causes damage to endothelial cells, the decreased state of activation of neutrophils during quiescent disease may be a beneficial effect of the treatment given to these patients.
In conclusion, in ANCA-associated vasculitides circulating neutrophils are not fully activated intravascularly, suggesting that a Shwartzman-like phenomenon may not primarily underlie the development of vascular damage. The increased expression of activation markers, such as CD66b, CD63 and CD64, on circulating neutrophils in patients with active vasculitis can be attributed to a state of pre-activation (‘priming’). These findings are consistent with the hypothesis that ‘primed’ neutrophils adhere to the endothelium, are then fully activated by ANCA, and release lytic enzymes and oxygen radicals at the endothelial cell surface, resulting in endothelial cell lysis, and eventually vasculitis.
Acknowledgments
The authors wish to thank M. G. Huitema, W. W. Oost-Kort and I. Bouma for their technical assistance, Dr J. Zijlstra, and Dr C. F. A. Franssen for their clinical input, and Dr P. C. Limburg for his valuable advice. This study was performed with the aid of departmental funds.
References
- 1.Kallenberg CGM, Brouwer E, Weening JJ, Cohen Tervaert JW. Anti-neutrophil cytoplasmic antibodies: current diagnostic and pathophysiological potential. Kidney Int. 1994;46:1–15. doi: 10.1038/ki.1994.239. [DOI] [PubMed] [Google Scholar]
- 2.Jennette JC, Falk RJ. Small vessel vasculitis. N Engl J Med. 1997;337:1512–23. doi: 10.1056/NEJM199711203372106. [DOI] [PubMed] [Google Scholar]
- 3.Cohen Tervaert JW, Mulder L, Stegeman C, et al. Occurrence of autoantibodies to human leucocyte elastase in Wegener's granulomatosis and other inflammatory disorders. Ann Rheum Dis. 1993;52:115–120. doi: 10.1136/ard.52.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charles LA, Falk RJ, Jennette JC. Reactivity of antineutrophil cytoplasmic autoantibodies with mononuclear phagocytes. J Leuk Biol. 1992;51:65–68. doi: 10.1002/jlb.51.1.65. [DOI] [PubMed] [Google Scholar]
- 6.Keogan MT, Esnault VL, Green AJ, et al. Activation of normal neutrophils by anti-neutrophil cytoplasm antibodies. Clin Exp Immunol. 1992;90:228–34. doi: 10.1111/j.1365-2249.1992.tb07934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai KN, Leung JC, Rifkin I, Lockwood CM. Effect of anti-neutrophil cytoplasm autoantibodies on the intracellular calcium concentration of human neutrophils. Lab Invest. 1994;70:152–62. [PubMed] [Google Scholar]
- 8.Mulder AH, Stegeman CA, Kallenberg CGM. Activation of granulocytes by anti-neutrophil cytoplasmic antibodies (ANCA) in Wegener's granulomatosis: a predominant role for the IgG3 subclass of ANCA. Clin Exp Immunol. 1995;101:227–32. doi: 10.1111/j.1365-2249.1995.tb08343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson PA, Alexander HD, McMillan SA, Maxwell AP. Up-regulation of the granulocyte adhesion molecule Mac-1 by autoantibodies in autoimmune vasculitis. Clin Exp Immunol. 1997;107:513–9. doi: 10.1046/j.1365-2249.1997.d01-956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 11.van Vollenhoven RF. Adhesion molecules, sex steroids, and the pathogenesis of vasculitis syndromes. Curr Opin Rheumatol. 1995;7:4–10. [PubMed] [Google Scholar]
- 12.Cohen Tervaert JW, Kallenberg CGM. The role of autoimmunity to myeloid lysosomal enzymes in the pathogenesis of vasculitis. In: Hansson GK, Libby P, editors. Immune functions of the vessel wall. Amsterdam: Harwood Academic Publishers; 1996. pp. 99–120. [Google Scholar]
- 13.Molad Y, Buyon J, Anderson DC, et al. Intravascular neutrophil activation in systemic lupus erythematosus (SLE): dissociation between increased expression of CD11b/CD18 and diminished expression of L-selectin on neutrophils from patients with active SLE. Clin Immunol Immunopathol. 1994;71:281–6. doi: 10.1006/clin.1994.1087. [DOI] [PubMed] [Google Scholar]
- 14.Argenbright LW, Barton RW. Interactions of leukocyte integrins with intercellular adhesion molecule 1 in the production of inflammatory vascular injury in vivo. The Shwartzman reaction revisited. J Clin Invest. 1992;89:259–72. doi: 10.1172/JCI115570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belmont HM, Buyon J, Giorno R, Abramson S. Up-regulation of endothelial cell adhesion molecules characterizes disease activity in systemic lupus erythematosus. The Shwartzman phenomenon revisited. Arthritis Rheum. 1994;37:376–83. doi: 10.1002/art.1780370311. [DOI] [PubMed] [Google Scholar]
- 16.Buyon JP, Shadick N, Berkman R, et al. Surface expression of Gp 165/95, the complement receptor CR3, as a marker of disease activity in systemic lupus erythematosus. Clin Immunol Immunopathol. 1988;46:141–9. doi: 10.1016/0090-1229(88)90014-1. [DOI] [PubMed] [Google Scholar]
- 17.Muller Kobold AC, Tulleken JE, Zijlstra JG, et al. Leukocyte activation in sepsis; correlations with disease state and mortality. 1998. Submitted. [DOI] [PubMed]
- 18.Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–92. doi: 10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 19.Stegeman CA, Cohen Tervaert JW, de Jong PE, Kallengerg CGM. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener's granulomatosis. N Engl J Med. 1996;335:16–20. doi: 10.1056/NEJM199607043350103. [DOI] [PubMed] [Google Scholar]
- 20.Cohen Tervaert JW, Huitema MG, Hene RJ, et al. Prevention of relapses in Wegener's granulomatosis by treatment based on antineutrophil cytoplasmic antibody titre. Lancet. 1990;336:709–11. doi: 10.1016/0140-6736(90)92205-v. [DOI] [PubMed] [Google Scholar]
- 21.Ziegler EJ, Fisher CJ Jr, Sprung CL, et al. Treatment of Gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. N Engl J Med. 1991;324:429–36. doi: 10.1056/NEJM199102143240701. [DOI] [PubMed] [Google Scholar]
- 22.Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM. 1994;87:671–8. [PubMed] [Google Scholar]
- 23.Exley AR, Bacon PA, Luqmani RA, et al. Development and initial validation of the Vasculitis Damage Index for the standardized clinical assessment of damage in the systemic vasculitides. Arthritis Rheum. 1997;40:371–80. doi: 10.1002/art.1780400222. [DOI] [PubMed] [Google Scholar]
- 24.Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–29. [PubMed] [Google Scholar]
- 25.Cohen Tervaert JW, Goldschmeding R, Elema JD, et al. Autoantibodies against myeloid lysosomal enzymes in crescentic glomerulonephritis. Kidney Int. 1990;37:799–806. doi: 10.1038/ki.1990.48. [DOI] [PubMed] [Google Scholar]
- 26.Mulder AH, Broekroelofs J, Horst G, et al. Anti-neutrophil cytoplasmic antibodies (ANCA) in inflammatory bowel disease: characterization and clinical correlates. Clin Exp Immunol. 1994;95:490–7. doi: 10.1111/j.1365-2249.1994.tb07024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuijpers TW, Tool AT, van der Schoot CE, et al. Membrane surface antigen expression on neutrophils: a reappraisal of the use of surface markers for neutrophil activation. Blood. 1991;78:1105–11. [PubMed] [Google Scholar]
- 28.Hamblin A, Taylor M, Bernhagen J, et al. A method of preparing blood leucocytes for flow cytometry which prevents upregulation of leucocyte integrins. J Immunol Methods. 1992;146:219–28. doi: 10.1016/0022-1759(92)90231-h. [DOI] [PubMed] [Google Scholar]
- 29.Muller Kobold AC, Kallenberg CGM, Cohen Tervaert JW. Leukocyte membrane expression of proteinase 3 correlates with disease activity in patients with Wegener's granulomatosis. Br J Rheumatol. 1998;37:901–7. doi: 10.1093/rheumatology/37.8.901. [DOI] [PubMed] [Google Scholar]
- 30.Sladek TL, Jacobberger JW. Flow cytometric titration of retroviral expression vectors: comparison of methods for analysis of immunofluorescence histograms derived from cells expressing low antigen levels. Cytometry. 1993;14:23–31. doi: 10.1002/cyto.990140106. [DOI] [PubMed] [Google Scholar]
- 31.Schwarz A, Fernandez-Repollet E. Development of clinical standards for flow cytometry. Ann N Y Acad Sci. 1993;677:28–39. doi: 10.1111/j.1749-6632.1993.tb38760.x. [DOI] [PubMed] [Google Scholar]
- 32.Lundahl J, Hed J. Differences in altered expression of L-selectin and Mac-1 in monocytes and neutrophils. Inflammation. 1994;18:67–76. doi: 10.1007/BF01534599. [DOI] [PubMed] [Google Scholar]
- 33.Schleiffenbaum B, Spertini O, Tedder TF. Soluble L-selectin is present in human plasma at high levels and retains functional activity. J Cell Biol. 1992;119:229–38. doi: 10.1083/jcb.119.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wayner EA, Garcia Pardo A, Humphries MJ, et al. Identification and characterization of the T lymphocyte adhesion receptor for an alternative cell attachment domain (CS-1) in plasma fibronectin. J Cell Biol. 1989;109:1321–30. doi: 10.1083/jcb.109.3.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinashi T, Springer TA. Adhesion molecules in hematopoietic cells. Blood Cells. 1994;20:25–44. [PubMed] [Google Scholar]
- 36.Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–34. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 37.Elsner J, Sach M, Knopf HP, et al. Synthesis and surface expression of ICAM-1 in polymorphonuclear neutrophilic leukocytes in normal subjects and during inflammatory disease. Immunobiology. 1995;193:456–64. doi: 10.1016/s0171-2985(11)80430-4. [DOI] [PubMed] [Google Scholar]
- 38.Metzelaar MJ, Wijngaard PL, Peters PJ, et al. CD63 antigen. A novel lysosomal membrane glycoprotein, cloned by a screening procedure for intracellular antigens in eukaryotic cells. J Biol Chem. 1991;266:3239–45. [PubMed] [Google Scholar]
- 39.Kuroki M, Yamanaka T, Matsuo Y, et al. Immunochemical analysis of carcinoembryonic antigen (CEA)-related antigens differentially localized in intracellular granules of human neutrophils. Immunol Invest. 1995;24:829–43. doi: 10.3109/08820139509060710. [DOI] [PubMed] [Google Scholar]
- 40.Ducker TP, Skubitz KM. Subcellular localization of CD66, CD67, and NCA in human neutrophils. J Leukoc Biol. 1992;52:11–16. doi: 10.1002/jlb.52.1.11. [DOI] [PubMed] [Google Scholar]
- 41.Valerius T, Repp R, de Wit TP, et al. Involvement of the high-affinity receptor for IgG (Fc gamma RI; CD64) in enhanced tumor cell cytotoxicity of neutrophils during granulocyte colony-stimulating factor therapy. Blood. 1993;82:931–39. [PubMed] [Google Scholar]
- 42.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–76. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 43.Riecken B, Gutfleisch J, Schlesier M, Peter HH. Impaired granulocyte oxidative burst and decreased expression of leucocyte adhesion molecule-1 (LAM-1) in patients with Wegener's granulomatosis. Clin Exp Immunol. 1994;96:43–47. doi: 10.1111/j.1365-2249.1994.tb06227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Macey MG, Jiang XP, Veys P, McCarthy D, Newland AC. Expression of functional antigens on neutrophils. Effects of preparation. J Immunol Methods. 1992;149:37–42. doi: 10.1016/s0022-1759(12)80046-9. [DOI] [PubMed] [Google Scholar]
- 45.Stibenz D, Buhrer C. Down-regulation of L-selectin surface expression by various leukocyte isolation procedures. Scand J Immunol. 1994;39:59–63. [PubMed] [Google Scholar]
- 46.Haller H, Eichhorn J, Pieper K, Goebel U, Luft FC. Circulating leukocyte integrin expression in Wegener's granulomatosis. J Am Soc Nephrol. 1996;7:40–48. doi: 10.1681/ASN.V7140. [DOI] [PubMed] [Google Scholar]
- 47.Reumaux D, Vossebeld PJ, Roos D, Verhoeven AJ. Effect of tumor necrosis factor-induced integrin activation on Fc gamma receptor II-mediated signal transduction: relevance for activation of neutrophils by anti-proteinase 3 or anti-myeloperoxidase antibodies. Blood. 1995;86:3189–95. [PubMed] [Google Scholar]
- 48.Charles LA, Caldas ML, Falk RJ, Terrell RS, Jennette JC. Antibodies against granule proteins activate neutrophils in vitro. J Leukoc Biol. 1991;50:539–46. doi: 10.1002/jlb.50.6.539. [DOI] [PubMed] [Google Scholar]
- 49.Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol. 1994;95:244–50. doi: 10.1111/j.1365-2249.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]