

Abstract
Numerous studies suggest that C-ANCA are directly pathogenic in vasculitis by activating leucocytes (oxidative burst, enzyme release, endothelial cytotoxicity, etc.). We and others have shown that C-ANCA can also directly activate HUVEC, but the precise target on HUVEC is unknown. We show in this study that C-ANCA recognize various targets on the HUVEC membrane (different from PR3 in our model), leading to secondary cell activation. Polyclonal affinity-purified C-ANCA recognized targets on the unfixed endothelial membrane in fluorescent ELISA, flow cytometry, and immunoprecipitation studies. C-ANCA did not react with Fcγ receptors. Reverse transcriptase-polymerase chain reaction (RT-PCR) experiments showed that HUVEC did not express PR3. The targets of polyclonal and monoclonal anti-PR3 antibodies on the endothelial membrane were not the same. Some epitopes were lost after trypsin–EDTA digestion and formaldehyde fixation of cells, whereas anti-PR3 targeted unfixed HUVEC. This suggests that anti-PR3 react with the endothelial membrane and recognize conformational epitopes shared with PR3. Endothelial cells may thus participate in the inflammation associated with Wegener's granulomatosis and contribute to the emergence of clinical manifestations.
Keywords: classic anti-neutrophil cytoplasmic antibodies, Wegener's granulomatosis, human umbilical vein endothelial cells
INTRODUCTION
The diagnostic and prognostic value of anti-neutrophil cytoplasmic antibodies (C-ANCA, or anti-PR3 antibodies) is well documented in Wegener's granulomatosis (WG) [1,2]. In vitro studies and animal models [3,4] suggest that C-ANCA could have a pathogenic role by inducing inflammatory reactions. The lesions observed in this disease may be secondary to leucocyte activation [5,6] after tumour necrosis factor-alpha (TNF-α) priming. ANCA activate polymorphonuclear cells through the FcγRII [7–10], leading to enzyme release, oxidative burst and endothelial damage [11]. ANCA are thought to activate other cell populations [12], such as endothelial cells [13,14] and monocytes, leading to the release of cytokines (e.g. IL-8). We and others have shown that C-ANCA activate endothelial cells and lead to endothelial wall expression of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), ELAM-1 and vascular cell adhesion molecule-1 (VCAM-1), as well as tissue factor, the main initiator of the coagulation cascade [13–16]. This effect results from direct endothelial stimulation by anti-PR3 antibodies, disappears when C-ANCA are neutralized, does not require other blood components, seems to be independent of circulating proinflammatory cytokines, and appears to involve at least local synthesis of IL-1α in the response to anti-PR3 [16].
The aim of this study was to identify the target(s) of C-ANCA on the endothelial membrane. Some authors have reported that cytokine-activated HUVEC express PR3, the C-ANCA target, leading to endothelial activation, while others argue that cytokine-stimulated HUVEC do not express PR3 [17,18].
MATERIALS AND METHODS
Reagents
Hanks' balanced salt solution (HBSS), fungizone, trypsin–EDTA, and streptomycin were from Gibco (Strasbourg, France). Colimycin (106 U/ml) was from Roger-Bellon (Neuilly sur Seine, France). Collagenase (CLS-1) was from Worthington Biochemical Co. (Freehold, NJ). Culture medium (M199 with Earl's salts) was from Eurobio (Les Ulis, France). Lymphoprep was from Nicomed (Oslo, Norway), lipopolysaccharide (LPS), ATPNa2, Triton X-100, orthodianisidine, PMSF, EDTA, EGTA, Trizma–bovine serum albumin (BSA) buffer (Trizma 50 mm, NaCl 0.1 m, BSA 1 mg/ml, pH 7.6), calcium chloride, octylglycoside (n-octyl β-D glucopyranoside), cycloheximide and polymyxin B were from Sigma (St Louis, MO). Fetal calf serum (FCS) was from Institut Jacques Bois (Reims, France). Human albumin (20%) was from Bio-Transfusion (Roissy, France). Dextran T 500, DEAE-Sephacel, S-200 Sephacryl, Hi-trap protein-G Sepharose, protein-A Sepharose beads and cyanogen bromide (CNBr)-activated Sepharose-4B were from Pharmacia (Uppsala, Sweden). Culture dishes were from Falcon (Polylabo, Strasbourg, France), and gelatin-coated plates were from Corning (Blois, France). The RNA polymerase chain reaction (PCR) GeneAmp kit was from Perkin Elmer (Roche, NJ). The RNA extraction RNA-plus kit was from Bioprobe Systems (Montreuil, France). The two sets of primers for PR3 were from Genset (Paris, France). ELISA tests were from Bio Advance (Emerainville, France) for anti-PR3 and anti-myeloperoxidase (MPO) antibodies, from Medgenix Diagnostics (SA Fleurus, Belgium) for TNF-α, and from Chromogenix AB for endotoxin. Human IgG Fc fragments, control IgG1, goat anti-human IgG and IgM and goat anti-human IgG, IgA and IgM–FITC-labelled antibodies were from Immunotech (Marseille, France). Anti-CD32 antibodies were from Serotec (Pantin, France). The anti-PR3 MoAb (WG2) was a kind gift from Dr E. Csernok (Medizinische Universität, Lübeck, Germany); anti-PR3 CLB-ANCA was from the German Red Cross.
Patients and sera
Six patients with biopsy-proven WG (ACR criteria [19]) were studied. All had active disease and multiorgan involvement, consisting of kidney disease (6/6), lung disease (4/6), skin disease (3/6), rheumatic disease (5/6), or ear, nose, throat (ENT) involvement (6/6). All had at least one positive biopsy (kidney and/or lung and/or nose) with typical features of WG. Patients were tested for ANCA by indirect immunofluorescent assay (IIF) and ELISA. Despite aggressive therapy, patients 3–5 died within 3 months of blood sampling. Sera were decomplemented (56°C, 45 min) and filtered, and immunoglobulins were purified with Hi-trap protein-G Sepharose. Decomplementation did not modify autoantibody recognition of PR3. Normal control sera (n = 20) were obtained from healthy blood donors. A pool of all the normal sera was used in the experiments, and immunoglobulin was extracted on Hi-trap protein-G Sepharose column and pooled (500 μg/ml). All preparations were tested for endotoxin contamination in the limulus amoebocyte assay and were used only if the concentration was < 0.05 ng/ml.
Affinity purification of anti-PR3 antibodies
Azurophilic granule proteins from polymorphonuclear neutrophils (PMN) were extracted by nitrogen cavitation (Parr Intrument, Moline, CA) and Percoll gradient sedimentation [20], as described elsewhere [15,16]. Briefly, azurophilic granules were isolated and sonicated on ice. Material containing PR3 was dialysed against equilibration buffer and applied to a DEAE-Sephacel column. Bound material was eluted with elution buffer. PR3 was present in the first peak. Fractions were pooled and dialysed overnight in 0.05 m CH3COONa pH 4.5, 0.05 m NaCl, 1% NaN3 and then applied to an S-200 column. Fractions were tested for PR3 and concentrated. SDS–PAGE showed only one protein band at 29 kD, the molecular weight of PR3. PR3 was quantified by weighing and ELISA. ELISA tests with specific antibodies against other granule components were negative. Purified PR3, identified by ELISA and SDS–PAGE (Fig. 1), was then coupled to CNBr-activated Sepharose 4B following the manufacturer's instructions. After dialysis against PBS, immunoglobulin was applied to the PR3–CNBr column and C-ANCA were eluted (PBS, 1 m NaCl, 1% NaN3), pooled and concentrated (500 μg/ml). Imunoglobulin fractions were free of PR3.
Fig. 1.
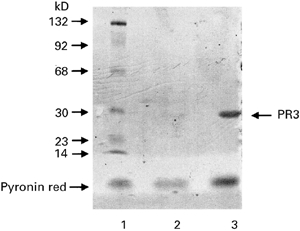
Silver staining of purified PR3. Proteins extracted on a DEAE–Sephacel column and a S-200 chromatography column were tested by SDS–PAGE analysis, which showed only one protein band at 29 kD, the molecular weight of PR3. Lane 1, molecular weight markers; lane 2, proteins extracted from the S2OO column; lane 3, proteins extracted from the DEAE column.
HUVEC preparation
Cells were harvested from human umbilical cord veins with 0.05% collagenase and grown to confluence in Bactogelatin-coated Petri dishes containing M199 culture medium supplemented with FCS 20%, l-glutamine 2 mm, penicillin 50 U/ml, streptomycin 50 μg/ml, and fungizone 5 μg/ml at 37°C in a 5% CO2 incubator. The medium was changed every 2 days until confluence. Only tightly confluent monolayers of secondary cultures (second passage) were used. HUVEC were identified by their morphology (cobblestone aspect in contrast-phase light microscopy) and their release of von Willebrand factor (vWF). Cell viability (trypan blue exclusion) was > 96%.
Cell stimulation
Endothelial cells were stimulated with various cytokines alone or in combination (IL-1 = 10 U/ml, TNF-α = 200 U/ml and interferon-gamma (IFN-γ) = 200 U/ml) for 1 h or 4 h. The cytokine combinations were as follows: IL-1 + TNF-α, IL-1 + IFN-γ, TNF-α+ IFN-γ, and IL-1 + TNF-α+ IFN-γ. Negative controls were unstimulated HUVEC, and positive controls were HL-60 cells.
RNA isolation and reverse transcriptase-PCR
Total RNA was isolated from 106 HUVEC using the RNA-Plus reagent according to the manufacturer's instructions. Total RNA (1 μg) was reverse-transcribed using the GeneAmp kit according to the manufacturer's protocol on a Biometra Trio-Thermoblock apparatus. Reverse transcription (RT) was followed by PCR amplification for 35 cycles in a volume of 100 μl. Two sets of primers for PR3 were used. The first was described by Mayet et al. [17], and the sequences of the sense and antisense primers were 5′-ATGGCC TCCCTGCAGATGCGGGGG-3′ and 5′-CGGAGG CACTGAGGTTGGCTGGGC-3′, respectively. The second set of primers was obtained from PR3 DNA (GenBank) using Oligo 4 software to optimize the various parameters and PCR conditions. Sequences for sense and antisense primers were 5′-GCACCTT GATCCACCCCAGCT-3′ and 5′-CGACGGAGGCACTGAGGT TGG-3′, respectively. The reaction cycle for PR3 (first set of primers) consisted of 94°C for 1 min, then 35 cycles of 50°C for 50 s and 70°C for 45 s; the reaction ended with a step at 70°C for 7 min and a waiting step at 4°C. The reaction cycle for PR3 using the second set of primers consisted of 94°C for 1 min, then 35 cycles composed of 50°C for 50 s and 64°C for 45 s; the reaction ended with a step at 70°C for 7 min and a waiting step at 4°C. Amplification products were electrophoresed on 2% agarose gel stained with ethidium bromide and photographed with Polaroid 665 negative films. RNA was quantified by scanning the bands with a Preference-Sebia densitometer.
Endothelial fixation of C-ANCA
Confluent HUVEC in 24-well plates were cultured with an excess of human normal serum or with anti-Fcγ antibodies to saturate FcγRII, then maintained at 4°C and gently agitated. Cells were incubated with known amounts of anti-PR3 antibodies. The supernatant was recovered at 0, 4 and 12 h and anti-PR3 were tested by ELISA. Endothelial fixation of anti-PR3 antibodies was also determined using flow cytometry analysis and direct fluorescent ELISA.
Flow cytometry
HUVEC were harvested from the dishes by mechanical scraping with a rubber policeman or by trypsin–EDTA digestion. Cells (500 000 cells/ml) were then washed three times in PBS with 10% human sera or Fc fragments from human immunoglobulin. The cells were then incubated with anti-PR3 antibodies (100 μg/ml) and gently agitated in an incubator for 1 h. Then cells were washed again in PBS and incubated for 30 min with an FITC-conjugated anti-human antibody. Cells were analysed on a FACScan flow cytometer (Becton Dickinson, Paris, France).
Fluorescent ELISA
Endothelial fixation of C-ANCA was determined by means of direct fluorescent ELISA on 96-well Bactogelatin-coated plates with culture medium containing 10% human serum. After washing with HBSS, cells were incubated with immunoglobulin from healthy subjects or with affinity-purified C-ANCA for 4 h at 4°C. After three further washing steps an FITC-labelled goat anti-human IgG was added and incubated for 1 h at 4°C. After further washes to remove unbound antibodies, plates were directly read at 405 nm on a Fluorescent ELISA plate reader (Biolumin; Molecular Dynamics, St Denis, France). Negative controls were unstimulated HUVEC (incubated or not with FITC-conjugated antibody) and HUVEC incubated with C-ANCA but not with the FITC-conjugated antibody. Normal controls were immunoglobulin extracted from a pool of 20 normal sera.
Endothelial cell membrane extracts
HUVEC membrane extracts were prepared using the method described by Hill et al. [21], with minor modifications. Briefly, 40 × 106 cells were harvested with a rubber policeman and placed in extraction buffer (Tris–HCl 200 mm pH 7.4, benzamidine 10 μg/ml, pepstatin 10 μg/ml, leupeptin 10 μg/ml, aprotinine 10 μg/ml, PMSF 100 μm, EDTA 0.5 mm, EGTA 0.5 mm, sucrose 857 mg, made up to 10 ml with water). Then cells were sonicated on ice (10 s, three times) and ultracentrifuged for 1 h at 105 g, 4°C. The pellet containing the lysed cell membranes was resuspended in immunoprecipitation buffer (Tris–HCl 10 mm (1200 ml), EDTA 5 mm (3 ml), NaCl 50 mm (1 ml), PMSF 1 mm (300 μl), H2O 24.2 ml, 10% Trition X-100 300 μl) for 3 h on ice, then sonicated and ultracentrifuged again; the supernatant containing the membrane proteins was concentrated and frozen.
Immunoprecipitation
HUVEC membrane proteins were incubated with anti-PR3 (affinity-purified or MoAbs, 100 μg/ml) for 3 h at 4°C on a wheel. Then 300 μl of Protein-A Sepharose beads washed twice in immunoprecipitation buffer were added and incubated on a wheel at 4°C for 3 h. The preparation was centrifuged for 5 min at 2500 g, and the pellet was collected and resuspended in 80 μl of 1× Laemmli buffer for 1 h. Aliquots were boiled for 7 min and centrifuged again to remove the beads. SDS–PAGE of 40-μg aliquots, together with molecular weight markers (14–205 kD), was performed using 10% bis cross-linked gels for 140 V per hour. Liquid electroblotting for 165 V per hour was used to transfer proteins to nitrocellulose membranes blocked with 1% PBS–Tween 40, 1% BSA. The membranes were incubated overnight with affinity-purified (or monoclonal) anti-PR3 antibodies diluted in PBS–Tween–BSA (1:1000). After four washes in PBS–Tween, membranes were incubated for 2 h with a peroxidase-conjugated goat anti-human IgG/A/M. After four further washes in PBS–Tween, the bound peroxidase conjugate was detected with a final 1-min incubation with luminol; chemiluminescence was measured with Fuji-RX x-ray films.
Anti-endothelial cell antibody testing
Sera from WG patients were tested for anti-endothelial cell antibodies (AECA) using a previously described method [22]. Briefly, HUVEC at the second passage were cultured to confluence, fixed in glutaraldehyde 0.1 and saturated in PBS–Tween–10% goat serum. Sera were used diluted 1:50 in PBS–Tween–10% goat serum. Bound immunoglobulins were revealed by adding peroxidase-labelled goat immune serum against either human IgG or IgM. A colour reaction was generated and plates were read at 492 nm. Results were expressed as arbitrary units by comparison with a reference curve obtained by including, in each microtitration plate, a positive control serum (from a patient with systemic lupus erythematosus and secondary anti-phospholipid syndrome) in two-fold dilutions starting at 1:25. The upper limit of normal was defined as the mean optical density (OD) + 3 s.d. for 103 sera from normal individuals.
Statistical analysis
Results are expressed as means ± s.d. and each value is the mean of at least three experiments performed in duplicate or in triplicate in different culture plates. Statwork software was used to calculate means, standard deviations (mean ± s.d.), and correlation parameters. Student's t-test was used to identify statistically significant differences (P < 0.05). Non-parametric tests were used in other cases.
RESULTS
Patients' sera and immunoglobulin
All six WG patients had a C-ANCA pattern (IIF assay, titre 1:400 to 1:800) and anti-PR3 antibodies (ELISA); screening for other autoantibodies was negative. Normal sera were devoid of anti-PR3 antibodies. Cytokine levels were determined in sera and immunoglobulin was used for stimulation. All but two sera were negative for TNF-α (patients 3 and 4: mean 6 pg/ml). IL-lα was detected in three WG sera (mean 2 ± 0.6 pg/ml). Screening for IL-1β was negative. Affinity-purified anti-PR3 and immunoglobulin from normal sera were devoid of cytokines (ELISA for TNF-α, IL-1α and IL-1β). Endothelial cells were always incubated with ANCA in the presence of polymyxin B (LPS inhibitor) at a final concentration of 2 μg/ml, even though endotoxin contamination of sera, affinity-purified immunoglobulin, culture medium and buffers was < 0.05 ng/ml. Purified anti-PR3 antibody fractions were devoid of PR3.
AECA testing
The sera and affinity-purified anti-PR3 from the six WG patients were tested for AECA. Neither IgG nor IgM-AECA was detected in the six WG patients. In contrast, AECA were detected in 50% of sera from lupus patients and in 90% of sera from scleroderma patients (data not shown).
Endothelial fixation of C-ANCA
There was a time-related decrease in the level of C-ANCA detected by ELISA in the supernatant (data not shown), supporting the hypothesis that C-ANCA fix to the endothelial membrane in a time-dependent manner, and not by FcγRII capture. The results were not modified when cells were preincubated with a blocking anti-FcγRII MoAb.
Fluorescent-ELISA
As shown in Table 1, a significantly stronger fluorescent response was obtained in C-ANCA-stimulated wells than in other wells (P = 0.001, Student's t-test), showing that C-ANCA fixed to endothelial cells while normal antibodies did not. Preincubation of ANCA with PR3 suppressed the fixation.
Table 1.
Fluorescent cyto-ELISA (see Materials and Methods)
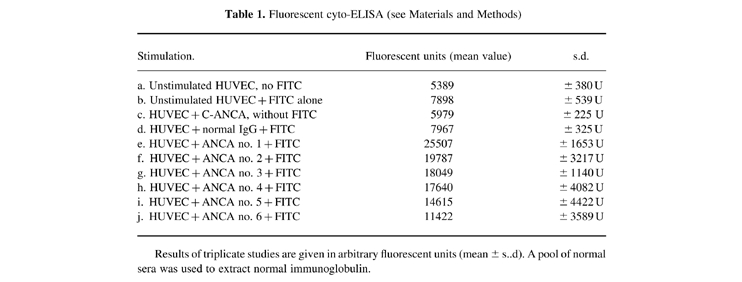
Results of triplicate studies are given in arbitrary fluorescent units (mean ± s..d). A pool of normal sera was used to extract normal immunoglobulin.
Flow cytometry
Controls consisted of unstimulated cells, cells incubated with normal IgG, and cells only incubated with the FITC-conjugated antibody. Cells harvested with trypsin-EDTA gave an inconsistent and very weak signal. Cells harvested by mechanical scraping showed a stronger signal (Fig. 2), which was present on all cells incubated with (mono or polyclonal) C-ANCA, but not on other cells (unstimulated or incubated with normal IgG). These results suggest that conformational epitopes on the HUVEC membrane were present after mechanical scraping but were removed by enzymatic digestion.
Fig. 2.
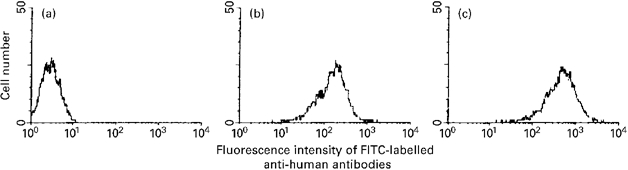
Flow cytometry. (a) Mean fluorescence intensity of HUVEC incubated with normal immunoglobulin and FITC-labelled anti-human antibodies. (b,c) Mean fluorescent intensity of HUVEC incubated with polyclonal C-ANCA isolated from a Wegener's granulomatosis (WG) patient (b) or with monoclonal anti-PR3 (c).
PR3 mRNA detection in endothelial cells
RT-PCR was performed on unstimulated cells, cytokine-stimulated cells and unstimulated HL-60 cells. Whatever the conditions of stimulation and primer sets, PR3-mRNA was never detected in HUVEC. As shown in Fig. 3, the only cells which expressed PR3 mRNA were HL-60 cells. PR3 transcripts were readily detected at the expected bands of 285 bp and 310 bp. To ensure that all samples contained similar amounts of cDNA, the same amount of RNA (1 μg) isolated from 106 endothelial cells was used for reverse transcription. Control amplification of the same RNA with glyceraldehyde-3-phosphate dehydrogenase (G3PDH) primers confirmed that equal amounts of RNA had been reverse transcribed. Cytokine stimulation induced no PR3-mRNA transcripts after 1 or 4 h. First-passage HUVEC did not contain PR3 mRNA (data not shown). As our endothelial cells did not express PR3 mRNA, PR3 was unlikely to be the target of C-ANCA in our model.
Fig. 3.
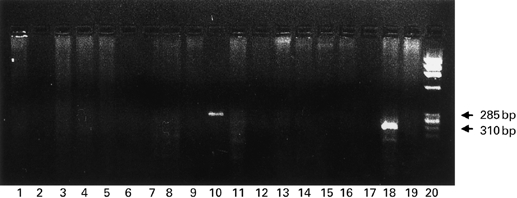
Reverse transcriptase-polymerase chain reaction (RT-PCR) for PR3. Lanes 1–9, first set of primers; lanes 11–19, second set of primers; lane 20, DNA scale. Negative control lane 1. HL-60 cells lanes 10 and 18. Cytokine-stimulated HUVEC lanes 2–9 and 11–17. Endothelial cells were stimulated with cytokines alone (IL-1, tumour necrosis factor-alpha (TNF-α), IFN-γ; lanes 1, 2, 3) or in various combinations (IL-1 + TNF-α, IL-1 + IFN-γ, TNF-α + IFN-γ, IL-1 + TNF-α + IFN-γ, or IL-1 + TNF-α (300 U) + IFN-γ; lanes 4, 5, 6, 7, 8). The same combinations were used in lanes 11–19. Only HL-60 cells expressed PR3 mRNA, transcripts being detected as the expected bands of 285 bp and 310 bp.
Immunoprecipitation and endothelial cell membrane extracts
HUVEC membrane extracts and immunoprecipitation experiments were carried out using either monoclonal anti-PR3 antibodies (WG2 and CLB-ANCA) or affinity-purified anti-PR3 from WG patients, as described in Materials and Methods. Figures 4 and 5 show typical immunoprecipitation results obtained with mechanically scraped HUVEC. ANCA recognized various epitopes on the HUVEC membrane, whereas normal sera did not react with the HUVEC membrane extract (data not shown). When HUVEC harvested with trypsin-EDTA were used, monoclonal (Fig. 4) and polyclonal (Fig. 5) anti-PR3 antibodies failed to react with any of the proteins on the nitrocellulose membrane, whereas the transfer was good as stated by Ponceau staining, strengthening the hypothesis that the C-ANCA target on the HUVEC membrane is a conformational epitope. Both of the monoclonal antibodies recognized the same endothelial targets (70, 92 and 132 kD and a faint band around 105 kD), whereas patients' polyclonal C-ANCA recognized different targets (82 kD in three cases, around 110 and 125 kD in the remainder).
Fig. 4.
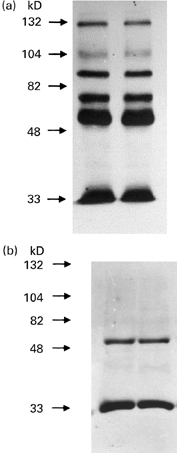
Immunoprecipitation of HUVEC membrane-bound proteins with murine monoclonal C-ANCA. (a) Immunoprecipitation of HUVEC membrane-bound proteins with two different monoclonal C-ANCA (lanes 1 (WG2) and 2 (CLB-ANCA)). (b) Control immunoprecipitation with the monoclonal C-ANCAs (lanes 1 (WG2) and 2 (CLB-ANCA)), using only the second antibody (peroxidase-conjugated).
Fig. 5.
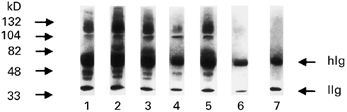
Immunoprecipitation of HUVEC membrane-bound proteins with affinity-purified polyclonal human C-ANCA (lanes 1–6) and IgG purified from normal control sera (lane 7).
DISCUSSION
Monoclonal C-ANCA and polyclonal affinity-purified C-ANCA from six patients with WG recognized targets on unfixed endothelial membranes in fluorescent-ELISA and flow cytometry. Affinity-purified polyclonal C-ANCA recognized several epitopes. The targets on the endothelial membrane differed for polyclonal and monoclonal anti-PR3 antibodies, pointing to different epitopes specificities. The observed effect was due to anti-PR3 antibodies and not to other antibodies, as shown by immuno-affinity purification and the absence of AECA. Interestingly, some epitopes were lost after trypsin-EDTA digestion of the HUVEC monolayer, as previously reported [23], and also after glutaraldehyde fixation, whereas anti-PR3 bound to unfixed HUVEC.
These results raise questions as to the precise target(s) of anti-PR3 on the endothelial cell membrane. Some authors have reported that PR3 is not expressed on unstimulated endothelial cells, but Mayet found that PR3 was expressed on HUVEC after TNF-α stimulation [17,18]. This is an elegant model. Other hypotheses should be considered. The first one is that PR3 may act as a ‘cofactor’ (as in the case of anti-β2-glycoprotein I (β2-GPI) antibodies): circulating PR3 (the target) may bind to HUVEC initially, followed by C-ANCA binding to PR3. This hypothesis cannot definitely be ruled out, as we used human sera for cell culture and binding site saturation, but it is very unlikely; moreover, this could play a role in vivo. Our purified ANCA did not contain PR3.
In our model ANCA did not react with Fcγ receptors. Moreover, as only cytokine-free culture conditions were used, PR3 should not have been expressed on HUVEC. Indeed, Mayet reported that PR3 expression by HUVEC required special culture conditions (extracellular matrix, stimulation by TNF-α and vascular endothelial growth factor (VEGF)) quite different from the conditions we used [24]. Using RT-PCR, we confirmed that HUVEC did not express PR3 in our model.
The epitopes recognized by C-ANCA on PR3 are not known, although one of them seems to be located on the NH2-terminal fragment [25]. Target epitopes on PR3 are conformational [26,27]. One hypothesis is that anti-PR3 reacts with the endothelial membrane by cross-reacting with membrane-bound proteins, as in other autoimmune diseases. For unknown reasons the HUVEC we used expressed targets that were recognized by affinity-purified anti-PR3 antibodies and by monoclonal anti-PR3, even though the HUVEC we used do not express PR3. The possibility that conformational epitope(s) were shared with PR3 and borne on the HUVEC membrane is valuable, as anti-PR3 antibodies recognized epitopes on the HUVEC membrane. As mouse MoAbs did not inhibit the binding of human polyclonal antibodies to HUVEC, and as targets recognized (immunoprecipitation) by the various antibodies (mono or polyclonal) were different, this relies on different epitopes specificities. Moreover, our results were obtained with HUVEC from neonatal large vessels and vasculitides occur in human adult microvasculature mature cells.
AECA are a class of autoantibodies detected in various clinical situations by their ability to bind to endothelial cells [22,23,28,29]. They seem to play a pathogenic role in some cases [30–32]. AECA have been detected in almost 50% of tested ANCA-containing sera [28,29]. There is no consensus on the best method to detect AECA [33,34]: some groups advocate the use of cell lines, while others use fixed or unfixed HUVEC [22,28–30]. Our results suggest that some C-ANCA could act as AECA.
Some AECA (like ANCA) seem to be pathogenic [28,29] but their targets are unknown [30,32,35]. AECA and ANCA can coexist in the same serum, all of them correlating with the disease activity and playing a pathogenic role [31,32]. In WG, AECA do not seem to be directed against FcγR, or against MHC class I or II molecules. Using radiolabelling of the cell surface and immunoprecipitation of the lysate, Meroni identified specific targets for AECA in WG, with one band at 130 kD and another at 170 kD [28,29]. This is not different from one of the bands recognized by the monoclonal C-ANCA. Our aim is now to purify and identify these endothelial targets.
Lastly, our results raise another problem, namely the detection of AECA and their isolation as a specific subgroup of autoantibodies. Antibodies directed against lysosomal enzymes of PMN (C-ANCA) can also target epitopes on the endothelial membrane, i.e. they play an ‘anti-endothelium activity’ or in other words are ‘anti-endothelial cell antibodies’. There is a need for these autoantibodies to be categorized and for the detection of AECA to be standardized [33,34].
In conclusion, our work gives new insights into ANCA-related diseases. Beside the activation of polymorphonuclear cells, leucocytes or monocytes, leading to enzyme release, oxidative burst and endothelial damage [5–11], it seems that ANCA activate endothelial cells [13–16], leading to modifications of endothelial functions (expression of adhesion molecules, synthesis of IL-1α…) possibly involved in the disease. The C-ANCA target in our HUVEC model is not PR3, suggesting that ANCA may initially first recognize endothelial targets, thereby inducing cell activation and cytokine synthesis and leading secondarily to the expression of membrane-bound PR3 that can be recognized by anti-PR3 antibodies.
Acknowledgments
This work was supported by INSERM and grants from La Société Française de Rhumatologie and Le Fond d'Études et de Recherche du Corps Médical des Hopitaux de Paris (AP-HP).
References
- 1.Gross WL, Schmitt WH, Csernok E. ANCA and associated diseases: immunodiagnostic and pathogenic aspects. Clin Exp Immunol. 1993;91:1–12. doi: 10.1111/j.1365-2249.1993.tb03345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kallenberg CGM, Brouwer E, Mulder HL, Stegeman CA, Weening JJ, Cohen Tervaert JW. ANCA pathophysiology revisited. Clin Exp Immunol. 1995;100:1–3. doi: 10.1111/j.1365-2249.1995.tb03594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gross WL, Csernok E, Flesch B. Classic anti-neutrophil cytoplasmic antibodies (C-ANCA), Wegener's autoantigen and their immunopathogenic role in Wegener's granulomatosis. J Autoimmun. 1993;6:171–84. doi: 10.1006/jaut.1993.1015. [DOI] [PubMed] [Google Scholar]
- 4.Tomer Y, Gilburd B, Blank M, et al. Characterization of biologically active anti-neutrophil cytoplasmic antibodies induced in mice. Arthritis Rheum. 1995;38:1375–81. doi: 10.1002/art.1780381004. [DOI] [PubMed] [Google Scholar]
- 5.Falk RJ, Terrell RS, Charles L, Jennette JC. Anti neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol. 1994;95:244–50. doi: 10.1111/j.1365-2249.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mulder HL, Stegeman C, Kallenberg CGM. Activation of granulocytes by anti-neutrophil cytoplasmic antibodies (ANCA) in Wegener's granulomatosis: a predominant role for the IgG3 subclass of ANCA. Clin Exp Immunol. 1995;101:227–32. doi: 10.1111/j.1365-2249.1995.tb08343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keogan M, Esnault VLM, Green A, Lockwood CM, Brown D. Activation of normal neutrophils by anti-neutrophil cytoplasm antibodies. Clin Exp Immunol. 1982;90:228–34. doi: 10.1111/j.1365-2249.1992.tb07934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brouwer E, Huitema M, Mulder LH, Heeringa P, van Goor H, Cohen-Tervaert JW, Weening JJ, Kallenberg CGM. Neutrophil activation in vitro and in vivo in Wegener's granulomatosis. Kidney Int. 1994;45:1120–31. doi: 10.1038/ki.1994.149. [DOI] [PubMed] [Google Scholar]
- 10.Porgess A, Recheda PB, Kimberly WT, Csernok E, Groos WL, Kimberly RP. ANCA engage and activate human neutrophils via Fcγ receptor RII. J Immunol. 1994;153:1271–80. [PubMed] [Google Scholar]
- 11.Mayet WJ, Schwarting A, Meyer Zum Buschenfelde K-H. Cytotoxic effects of antibodies to proteinase 3 (C-ANCA) on human endothelial cells. Clin Exp Immunol. 1994;97:458–65. doi: 10.1111/j.1365-2249.1994.tb06110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casselman B, Kilgore K, Miller B, Warren J. Antibodies to neutrophil cytoplasmic antigens induce monocyte chemoattractant protein-1 secretion from human monocytes. J Lab Clin Med. 1995;126:495–502. [PubMed] [Google Scholar]
- 13.Mayet WJ, Meyer Zum Buschenfeld K-H. Antibodies to proteinase 3 increase adhesion of neutrophils to human endothelial cells. Clin Exp Immunol. 1993;94:440–6. doi: 10.1111/j.1365-2249.1993.tb08215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayet WJ, Schwarting A, Orth T, Meyer Zum Büschenfeld K-H. Antibodies to proteinase 3 mediate expression of vascular cell adhesion molecule-1 (VCAM-1) Clin Exp Immunol. 1996;103:259–67. doi: 10.1046/j.1365-2249.1996.d01-626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Bandt M, Meyer O, Hakim J, Pasquier C. Antibodies to proteinase-3 mediate expression of intercellular adhesion molecule-1. Brit J Rheum. 1997;36:839–46. doi: 10.1093/rheumatology/36.8.839. [DOI] [PubMed] [Google Scholar]
- 16.De Bandt M, Ollivier V, Meyer O, Babin-Chevaye C, Khechaıamp;;uml; F, de Prost D, Hakim J, Pasquier C. Induction of interleukin-1 and subsequent tissue factor expression by anti-proteinase-3 antibodies in human umbilical vein endothelial cells. Arthritis Rheum. 1997;40:2030–8. doi: 10.1002/art.1780401116. [DOI] [PubMed] [Google Scholar]
- 17.Mayet WJ, Csernok E, Szymkoviak C, Groos WL, Meyer Zum Buschenfeld K-H. Human endothelial cells express proteinase 3, the target antigen of anticytoplasmic antibodies in Wegener's granulomatosis. Blood. 1993;82:1221–9. [PubMed] [Google Scholar]
- 18.King WJ, Adu D, Daha MR, Brooks CJ, Radford DJ, Pall AA, Savage COS. Endothelial cells and renal epithelial cells do not express the Wegener's autoantigen, proteinase 3. Clin Exp Immunol. 1995;102:98–105. doi: 10.1111/j.1365-2249.1995.tb06642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leavitt RY, Fauci AS, Bloch DA, et al. The American college of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. 1990;33:1101–7. doi: 10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 20.Wiik A, Rasmussen N, Wieslander J. Methods to detect auto-antibodies to neutrophilic granulocytes. In: Van Venrooij, editor. Manual of biological markers of diseases. A 9. Dordrecht: Kluwer Academic Publisher; 1993. pp. 1–14. [Google Scholar]
- 21.Hill BM, Phipps JL, Cartwight RJ, Milford-Ward A, Greaves M. Antibodies to membranes of endothelial cells and fibroblasts in scleroderma. Clin Exp Immunol. 1996;106:491–7. doi: 10.1046/j.1365-2249.1996.d01-867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer O, Kaiser P, Haim T, et al. Antivascular endothelial cell antibodies (AECA). Comparison of two assay methods and clinical applications. Rev Rhum (English ed.) 1995;11:737–43. [PubMed] [Google Scholar]
- 23.Cines DB, Lyss AP, Reeber M, et al. Presence of complement fixing antiendothelial cell antibodies in systemic lupus erythematosus. J Clin Invest. 1984;73:611–25. doi: 10.1172/JCI111251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faust J, Brockman H, Meyer K, Zum Buschenfelde K-H, Mayet WJ. Regulation of proteinase-3 mRNA-expression in endothelial and renal epithelial cells. Arthritis Rheum. 1997;40(Suppl. 9):220. Abstr. [Google Scholar]
- 25.Bini P, Gabay J, Teitel A, Melchior M, Zhou JL, Elkon KB. Antineutrophil cytoplasmic antibodies in Wegener's granulomatosis recognise conformational epitopes on proteinase 3. J Immunol. 1992;149:1409–15. [PubMed] [Google Scholar]
- 26.Specks U, Fass DN, Fautsch MP, Hummel AM, Wiss MA. Recombinant human proteinase-3, the Wegener's autoantigen, expressed in HMC-1 cells is enzymatically active and recognised by C-ANCA. FEBS Letters. 1996;390:265–70. doi: 10.1016/0014-5793(96)00669-2. [DOI] [PubMed] [Google Scholar]
- 27.Chang L, Binos S, Savige J. Epitope mapping of anti-proteinase 3 and anti-myeloperoxidase antibodies. Clin Exp Immunol. 1995;102:112–9. doi: 10.1111/j.1365-2249.1995.tb06644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savage COS, Pottinger BE, Gaskin G, Lockwood CM, Pusey CD, Pearson JD. Vascular damage in Wegener's granulomatosis and microscopic polyarteritis: presence of anti-endothelial cell antibodies and their relation to antineutrophil cytoplasm antibodies. Clin Exp Immunol. 1991;85:14–19. doi: 10.1111/j.1365-2249.1991.tb05675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meroni PL, Del Papa N, Conforti G, Barcellini W, Borghi M, Gambini D. Antibodies to endothelial cells in systemic vasculitis. In: Cervera R, Khamashta MA, Hughes GRV, editors. Antibodies to endothelial cells and vascular damage. London: CRC Press; 1994. pp. 121–32. [Google Scholar]
- 30.Carvalho D, Savage COS, Black C, Pearson JD. IgG anti-endothelial cell autoantibodies from scleroderma patients induce leukocyte adhesion to human vascular endothelial cells in vitro. J Clin Invest. 1996;97:111–9. doi: 10.1172/JCI118377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frampton G, Jayne DR, Perry G, Lockwood CM, Cameron JS. Antibodies to endothelial cells and neutrophil cytoplasmic antigen in systemic vasculitis. Clin Exp Immunol. 1990;82:227–32. doi: 10.1111/j.1365-2249.1990.tb05431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Del Papa N, Guidali L, Sironi M, et al. Anti-endothelial cell IgG antibodies from patients with Wegener's granulomatosis bind to human endothelial cells in vitro and induce adhesion molecules expression and cytokine secretion. Arthritis Rheum. 1996;39:758–66. doi: 10.1002/art.1780390507. [DOI] [PubMed] [Google Scholar]
- 33.Yiounou P, Meroni PL, Khamashta MA, Shoenfeld Y. A need for standardization of the anti-endothelial cell antibodies test. Immunol Today. 1996;16:363–4. doi: 10.1016/0167-5699(95)80001-8. [DOI] [PubMed] [Google Scholar]
- 34.Meroni P, Khamashta MA, Youinou P, Shoenfeld Y. Conference report mosaic of antiendothelial cell antibodies. Review of the first international workshop on anti-endothelial antibodies: clinical and pathological significance. Milan, 9 November 1994. Lupus. 1995;4:95–99. doi: 10.1177/096120339500400203. [DOI] [PubMed] [Google Scholar]
- 35.Saadi S, Holzknecht L, Patte CP, Stern DM, Platt JL. Complement-mediated regulation of tissue factor activity in endothelium. J Exp Med. 1995;182:1807–11. doi: 10.1084/jem.182.6.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]