

Abstract
Phagocyte and NK cell CR3 functions as both an adhesion molecule and an iC3b receptor mediating cytotoxic responses to microorganisms. Cytotoxic activation of iC3b receptor function requires ligation of both a CD11b I-domain site for iC3b and a lectin site located in the C-terminus of CD11b. Because tumours lack the CR3-binding polysaccharides of bacteria and fungi, iC3b-opsonized tumours do not stimulate CR3-dependent cytotoxicity. Previous studies showed that NK cells could be induced to kill iC3b-opsonized tumours with small soluble β-glucans that bound with high affinity to CR3, bypassing the absence of similar polysaccharides on tumour membranes. Because CR3 signalling requires several tyrosine phosphorylation events, it appeared possible that CR3-dependent killing of autologous tumour cells might be suppressed by NK cell inhibitory receptors for MHC class I (KIR and CD94/NKG2) whose action involves recruitment of SHP-1 and SHP-2 tyrosine phosphatases. In the current study, Epstein–Barr virus (EBV)-transformed B cells were used as targets following opsonization with iC3b. Soluble β-glucan primed CR3 for killing of iC3b-coated B cells, but autologous class I-bearing targets were 84% more resistant than class I-deficient Daudi cells. Blockade of target cell class I with a MoAb specific for a domain recognized by both KIR and CD94/NKG2 resulted in comparable killing of class I+ B cells. By contrast, another MoAb to class II had no effect on cytotoxicity. These data suggest that NK cell recognition of class I suppresses CR3/tyrosine kinase-dependent cytotoxicity in the same way as it suppresses cytotoxicity mediated by other tyrosine kinase-linked receptors such as FcγRIIIA (CD16).
Keywords: NK cells, complement, immunomodulators, adhesion molecules, tumour immunity
INTRODUCTION
The αMβ2-integrin known also as Mac-1, CR3, or CD11b/CD18 has two major functions on phagocytic cells. As the Mac-1 adhesion molecule it mediates the diapedesis of leucocytes through the endothelium via generation of a high-affinity binding site for intercellular adhesion molecule-1 (ICAM-1) [1–3]. As CR3, it mediates phagocytic and degranulation responses to microorganisms or immune complexes opsonized with iC3b [4–6]. For these functions, the Mac-1/CR3 molecule goes through a series of ‘inside-out’ and/or ‘outside-in’ signalling steps that result in exposure of high-affinity binding sites and/or altered linkage to the actin cytoskeleton. The nature of these activation and signalling pathways has not been completely defined, and it is particularly unknown whether activation for cytotoxic responses involves a similar pathway of events as the signalling for acquisition of the high-affinity ICAM-1 binding site and firm adhesion.
CR3 signalling for cytotoxic degranulation in response to iC3b-opsonized yeast requires ligation of both an iC3b-binding site in the I-domain of CD11b [7] and a separate lectin site that maps C-terminal to the I-domain [8,9]. Although iC3b-opsonized tumour cells cannot trigger CR3 cytotoxic activation because their membranes lack the CR3-binding polysaccharides of microbial cell walls, soluble β(1–3)-glucan polysaccharides isolated from fungi can bind to CR3 with high affinity and prime the receptor for subsequent cytotoxic activation by iC3b-tumour cells that are otherwise inert in stimulating CR3-dependent cytotoxicity [10,11]. Polysaccharide priming of CR3 for cytotoxicity of iC3b-target cells involves a Mg2+ and protein tyrosine kinase (PTK)-dependent conformational change in CD11b that exposes the activation epitope defined by MoAb CBRM1/5, but not the high-affinity ICAM-1 reporter epitope defined by MoAb 24 [10].
Following priming with soluble β-glucan, the CR3 of NK cells was shown to mediate the cytotoxicity of freshly excised breast tumour cells by ligation to opsonic iC3b that is present on most tumours cells as the result of IgM and IgG tumour-bound antibodies that activate the classical pathway of C [11]. However, because NK cells from allogeneic donors were used as effectors in that investigation, it remained possible that such cytotoxicity might not have occurred with the patients' own NK cells because of autologous MHC class I on tumour cells [12–14]. NK cells express two classes of inhibitory receptors that suppress cytotoxicity through recognition of homologous class I on target cells. Killer cell inhibitory receptors (KIR) belong to the immunoglobulin superfamily and the CD94/NKG2 receptors are disulphide-linked dimers belonging to the C-type lectin family [14]. Although some types of NK cell cytotoxic activation (e.g. killing of K562 cells) are not suppressed by these inhibitory receptors [15], the common mechanism of suppression used by both KIR and CD94/NKG2 that involves the recruitment of SHP-1 and SHP-2 tyrosine phosphatases to block activating receptors that signal through PTK [14,16] suggested that PTK-linked CR3 might be under similar class I control.
This study sought to determine if NK cell CR3-dependent cytotoxicity might be negatively regulated by recognition of target cell class I. The Burkitt's lymphoma-derived B cell line Daudi, a model class I-deficient tumour, was compared with an Epstein–Barr virus (EBV)-transformed B cell line derived from a donor whose NK cells were later tested for CR3-dependent cytotoxicity against both target cell types. A partial resistance of the autologous iC3b-target cells to CR3-dependent cytotoxicity was shown to be class I-dependent, suggesting that NK cell CR3 is regulated by KIR and/or CD94/NKG2 in the same manner as NK cell FcγRIIIA [16].
MATERIALS AND METHODS
B lymphoblastoid cell lines
Daudi B-lymphoblastoid cells and the 721.221 B cell line [17] were obtained from the American Type Culture Collection (ATCC, Rockville, MD). A B cell line was also derived from peripheral blood lymphocytes (PBL) of a normal volunteer by transformation with EBV [18]. Each cell line was maintained in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics.
Opsonization of B cell lines with C3
Harvested cultures were washed three times and suspended at 2.5 × 106/ml in warm RPMI 1640. Serum collected from a young male AB+ blood donor and stored frozen at −80°C was thawed rapidly at 37°C, maintained in an ice bath, and diluted with ice-cold RPMI 1640 just before use. Serum dilutions were mixed with an equal volume of B cell line suspension and incubated in a shaking water bath at 37°C for 45 min. The serum-opsonized cells were washed three times and resuspended at 5 × 106/ml in ice-cold RPMI 1640 culture medium. A portion of the cells was analysed for bound C3 fragments by staining with anti-C3–FITC and flow cytometry (see below), and the remaining cells were labelled for use as targets for NK cell cytotoxicity assays (see below).
Polyclonal antibodies and MoAbs
Rabbit antisera to human C3c and C3d were mixed together and used for generation of rabbit IgG anti-C3–FITC [11]. Rat MoAbs to C3c, C3g, and C3d and their use in determining the type and quantity of bound C3 fragments on sheep erythrocytes following coupling to 125I [19] were conjugated instead to FITC for analysis of bound C3 fragments on B cell lines using flow cytometry. A MoAb recognizing a neoepitope of bound iC3b (‘C3bi-neoantigen’) that is absent in bound C3b, C3dg, or C3d was purchased from Quidel (San Diego, CA) and coupled to FITC. The hybridoma line secreting the IgG1 MoAb 3D9 to CD35 (CR1) was a gift from Dr J. Atkinson (Washington University, St Louis, MO). The cell lines secreting the IgG2a MoAb OKM1 anti-CD11b (C-terminal domain), the IgG2a MoAb HB-5 to CD21 (CR2) and MOPC-21 myeloma IgG1 were obtained from ATCC. DX17, a mouse IgG1 MoAb to a human MHC class I framework epitope shared by HLA-A, -B, and -C [20] was the generous gift of Dr L. L. Lanier (DNAX Research Institute, Palo Alto, CA). MEM 136, an IgG1 anti-class II, was kindly provided by Dr V. Horejsí (Czech Academy of Sciences, Prague, Czech Republic). IgG fractions were isolated from ascites fluid by precipitation with ammonium sulphate followed by Mono-Q chromatography [21]. The anti-CD56 MoAb NKH1A [22] (kindly provided by Dr J. Ritz of Harvard Medical School, Boston, MA) and the MEM 188 anti-CD56 MoAb (kindly provided by Dr V. Horejsí) were each conjugated to FITC and used alternatively for identification of CD56+ NK cells. Goat F(ab′)2 anti-mouse IgG–FITC was purchased from Southern Biotechnology Associates (Birmingham, AL).
Immunofluorescence staining and flow cytometry
Fluorescence staining and flow cytometry were carried out using a Coulter Profile II instrument [11] followed by analysis of list mode data using the Pro2FCS and WinList 3.0 software packages from Verity Software House (Topsham, ME).
Isolation of soluble β-glucan
A low mol. wt polysaccharide fraction was isolated from zymosan by solubilization with hot formic acid [9]. There was some variation in the size and sugar composition of such soluble zymosan polysaccharide (SZP) preparations that was dependent upon the lot of zymosan. Analysis of various SZP preparations indicated a homogeneous sized polysaccharide of 10–20 kD made up of variable proportions of glucose and/or mannose. Some preparations of SZP were found to have no detectable mannose and to be made up entirely of β(1,3)-glucan, whereas other preparations were found to contain up to 90% mannose. As reported previously, such preparations of pure β-glucan exhibited comparable CR3-binding [9] and priming [10] activity to the other SZP preparations containing primarily mannose. The SZP preparation used in this study consisted almost entirely of β-glucan and had an estimated mol. wt of approx. 10 kD by S-200HR chromatography [9]. Accordingly, it is referred to as ‘SZP β-glucan.’ When this SZP was labelled with 125I [9], it exhibited saturation binding to neutrophil CR3 at concentrations of 2–4 μg hexose/ml that was blocked by either a 50–100-fold molar excess of unlabelled SZP or various MoAbs to CD11b [9].
NK cell isolation and cytotoxicity assays
NK cells were isolated using a ‘NK Cell Isolation Kit’ (Miltenyi Biotec, Auburn, CA). Briefly, mononuclear cells were isolated from citrated blood on a Histopaque density gradient. After five washes with RPMI 1640, the cells were incubated with a cocktail of modified antibodies specific for CD3, CD4, CD19 and CD33 to label non-NK cells for 15 min at 4°C. After three washes with RPMI 1640, the antibody-coated cells were incubated with colloidal super-paramagnetic MACS MicroBeads for an additional 15 min at 4°C. The cell suspension was passed through 30 μg of nylon mesh to remove clumps and applied to the top of a prefilled AS column. The eluted NK cells were washed four times in RPMI 1640 and assessed for NK cell purity by staining with anti-CD56–FITC and flow cytometry. The percentage of CD56+ NK cells in this population was ≥ 90%, and the viability was ≥ 92%.
Isolated NK cells (1 × 106/ml; 0.1 ml/well) in V-bottomed 96-well microplates (Dynatech Labs, Inc., Chantily, VA) were incubated with various activating or blocking agents (i.e. β-glucan, MoAb) for 30 min at 37°C and then washed three times with RPMI 1640. The following concentrations were used: 1.0 μg/ml of IgG MoAb or myeloma IgG; 2.0 μg/ml of SZP β-glucan. IgG preparations were centrifuged (14 000 g for 20 min at 4°C) just before addition to NK cells to remove small soluble aggregates that can inhibit cytotoxicity non-specifically via Fc receptors. For analysis of cytotoxicity, the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, Madison, WI) was used. After one wash with RPMI 1640, 100 μl of target cells (0.5–2.0 × 105/ml in RPMI 1640; giving E:T cell ratios of 5:1–20:1) were added to each well and the plate was incubated at 37°C for 4 h. The plate was centrifuged, and the absorbance at 490 nm was evaluated using an STL ELISA reader (Tecan U.S., Research Triangle Park, NC). Lytic units required for 20% lysis per 1 × 107 effector cells were calculated as described by Bryant et al. [23].
RESULTS
Generation of class I+ and class I − B cell targets opsonized with same amount of iC3b
EBV-transformed B cell lines have been used extensively to explore the nature of class I recognition by NK cells, and the class I-deficient 721.221 line has proved especially useful following stable transfection with various HLA-A, -B, or -C genes [15]. Unexpectedly, attempts to generate iC3b-opsonized 721.221 cells for use as targets for NK cell CR3-dependent cytotoxicity were unsuccessful. Although most B cell lines activate the alternative pathway of C [24–26], this requires the expression of CR2 [25,27,28], and CR2 was undetectable on 721.221 cells (not shown). Accordingly, Daudi cells were examined as possible targets, as Daudi cells lack membrane class I as the result of β2-microglobulin deficiency [29] and express amounts of CR2 sufficient to activate C [30]. An EBV-transformed class I+ B cell line generated from a normal volunteer was used so that paired autologous NK cells from this donor could be examined for lytic activity. Incubation of each cell line in serum from an AB+ donor diluted 1:4 resulted in deposition of C3 on both cell types, but far more C3 was detected on the Daudi cells with anti-C3–FITC (not shown). To produce C3 deposition on each cell line that was nearly equivalent, it was necessary to use serum diluted 1:10 with Daudi cells and 1:4 with the normal B cell-derived line (Fig. 1). Analysis of the relative size of individual cells in the two cell lines from forward scatter light plots indicated that the average cell from the EBV-transformed B cell line was ≤ 12% larger than the average Daudi cell (not shown). However, analysis of staining by anti-C3b–FITC and anti-C3g–FITC indicated that cells from the EBV-transformed B cell line also bore ≥ 15% more iC3b per cell than did the Daudi cells (Fig. 1), indicating that the actual iC3b density per cell was close to being equal when the slightly larger size of the EBV-transformed B cell line was taken into consideration. The reduced uptake of C3 by the normal B cell-derived line may have resulted because of its abundant CR1 expression (not shown) compared with the CR1− Daudi cells [31]. Further analysis of the two serum-opsonized cell lines with MoAbs to C3bi-neoantigen, C3c, C3g, and C3d indicated that the normal B cell-derived line bore slightly more C3dg than the Daudi cells (Fig. 1). Since CR1 serves as a cofactor for serum factor I proteolysis of fixed iC3b into fixed C3dg [32], this indicates that some of the fixed iC3b on the normal B cell line expressing CR1 was degraded to C3dg.
Fig. 1.
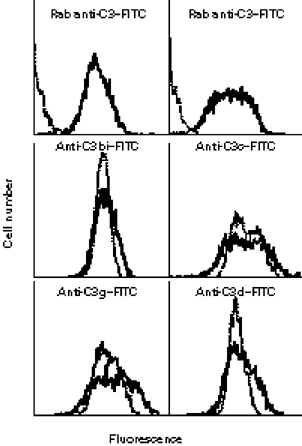
Analysis of C3 fragments deposited onto target B cell lines using flow cytometry and MoAbs to specific C3 epitopes. Staining was carried out with a rabbit anti-C3c/C3d–FITC mixture (Rab anti-C3–FITC) or FITC-labelled MoAbs specific for C3bi, C3c, C3g, and C3d. Unopsonized target cells were used to assess non-specific staining. The upper panels compare the normal B cell-derived line on the left and Daudi cells on the right, with the solid lines representing the serum-opsonized cells and the dotted lines representing the unopsonized cells. The remaining four panels compare staining only with the two serum-opsonized cell types, with the solid lines representing the normal B cell-derived targets and the dotted lines representing Daudi cells.
Sensitivity of iC3b-opsonized B cell lines to NK cell cytotoxicity
CD56+ NK cells were isolated from the same volunteer whose PBL had been used to generate the class I+ B cell line and tested for cytotoxicity of this autologous B cell line versus class I− Daudi cells No significant cytotoxicity of either B cell line occurred with unactivated NK cells, even after opsonization with iC3b/C3dg (Fig. 2, data are shown only for the iC3b-opsonized target cells, as there were virtually no lytic units of activity measurable with the unopsonized targets). As with other tumour cell types [10,11], the addition of 2 μg/ml SZP β-glucan also failed to induce cytotoxicity when the B cell targets lacked the CR3 target ligand iC3b (not shown). The cytotoxicity mediated by β-glucan priming with the iC3b-opsonized B cells was blocked by addition of OKM1 anti-CR3 (but not mouse myeloma IgG), confirming that such iC3b/β-glucan-induced cytotoxicity was CR3-dependent. Despite a coating with an equivalent cell surface density of iC3b, the autologous class I+ B cells were 84% more resistant to cytotoxicity than were the class I− Daudi cells.
Fig. 2.
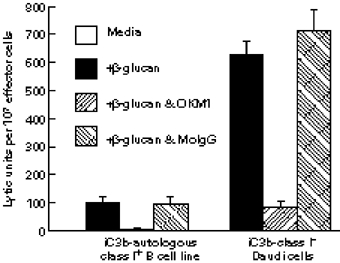
Reduced NK cell CR3-dependent cytotoxicity when an autologous class I+ Epstein–Barr virus (EBV)-transformed B cell line target is compared with class I− Daudi cells. NK cells were tested for cytotoxicity in a 4-h assay at E:T cell ratios ranging from 5:1 to 20:1 and the lytic units (for 20% cytotoxicity) per 107 effector NK cells were calculated. A B cell line generated from the NK cell donor's peripheral blood lymphocytes (PBL) was used as a target in comparison with class I− Daudi cells. Priming of NK cell CR3 by addition of 2 μg/ml of soluble β-glucan to the reaction medium resulted in 625 ± 60 lytic units with the iC3b Daudi cells, but only 100 ± 9 lytic units with the iC3b autologous B cell line. Killing was blocked by OKM1 anti-CR3 but not by non-specific mouse IgG (MoIgG). No lytic units of cytotoxicity were detected with either target cell type in the media control lacking β-glucan.
Blockade of target cell class I, but not class II, allows NK cells to mediate CR3-dependent cytotoxicity of autologous B cell lines with equal efficiency as with class I− Daudi cells
The reduced cytotoxicity of the autologous B cell line compared with Daudi cells could have been due to inherent differences between the two cell lines in lytic sensitivity (e.g. reduced sensitivity to perforin insertion; more efficient membrane repair) or it might have been due to a suppression of NK cell CR3 signalling brought about by NK cell recognition of autologous target cell class I. When the same NK cell cytotoxicity assays incorporated an IgG1 anti-HLA MoAb (DX17) that was known to block both the KIR and CD94/NKG2 recognition domains of class I molecules [33,34], β-glucan-induced NK cell killing of iC3b-opsonized autologous class I+ B cells was equivalent to the CR3-dependent cytotoxicity observed with iC3b-opsonized Daudi cells (Fig. 3). The four-fold increase in cytotoxicity (P ≤ 0.001) was CR3-dependent because it was blocked by a mixture of OKM1 anti-CR3 with DX17 anti-class I, and because little cytotoxicity was induced by the anti-class I without priming by β-glucan (not shown). As expected, the anti-class I MoAb had no effect on β-glucan-induced NK cell lysis of the class I− iC3b-opsonized Daudi cells. Furthermore, this absence of any enhancement of cytotoxicity by DX17 anti-class I with the Daudi target cells shows that the effect of the anti-class I was mediated by its attachment to target cell class I and not by its attachment to the class I of NK effector cells. It was also important to show that the mere coating of the target cells with IgG did not promote antibody-dependent cellular cytotoxicity. MEM 136, an IgG1 MoAb to class II, was used for this control, since each of the B cell targets had been shown to stain with the same fluorescence intensity using this MoAb followed by F(ab′)2 anti-mouse IgG–FITC (not shown). Neither MEM 136 nor MOPC-21 mouse myeloma IgG1 (that did not bind to the targets) caused a significant change in cytotoxicity of either iC3b-target (Fig. 3).
Fig. 3.
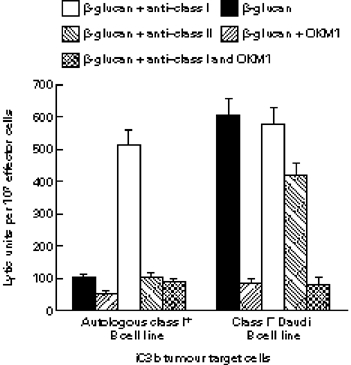
Enhancement of NK cell CR3-dependent cytotoxicity of autologous B cell line targets by blockade of target cell class I but not class II. A class I+ B cell line and class I− Daudi cells were opsonized with equivalent amounts of iC3b as in Fig. 1 and tested for lytic sensitivity to NK cells isolated from the same donor as the B cell line as described in Fig. 2 An IgG1 MoAb to class I (DX17) increased the β-glucan/CR3-dependent lytic sensitivity of the autologous B cell line up to the same level as Daudi cells, while an IgG1 MoAb to class II (MEM 136), had no significant effect on cytotoxicity of either target cell.
DISCUSSION
This study shows that PTK-linked NK cell CR3 are probably regulated by tyrosine phosphatase-linked receptors for class I (KIR and/or CD94/NKG2) in a manner that renders autologous class I-bearing tumour cells partially resistant to cytotoxicity. EBV-transformed B cell lines were used as tumour target cells following opsonization with iC3b. An autologous class I+ B cell line was 84% less sensitive to NK cell CR3-dependent cytotoxicity than were class I− Daudi cells, and blockade of target cell class I, but not class II, increased NK cell CR3-dependent cytotoxicity of the autologous target cells up to the same level as the class I− Daudi cells. Because class I only partially inhibited NK cell CR3-dependent cytotoxicity, and particularly also because tumour cell class I is usually lost as part of the metastatic process, tumours bearing naturally occurring or vaccine elicited antibody and C3 may be particularly susceptible to therapy with β-glucan.
B lymphoblastoid cell lines are known to activate the alternative pathway of C, resulting in deposition of iC3b that is a non-activating ligand for NK cell CR3 [24,27,28,30,35,36]. Although other tumour types may also activate the alternative pathway of C, opsonization of patient tumours with C3 frequently also occurs because of natural or tumour-elicited antibody and the classical pathway of C. Such antibodies in patients with mammary carcinoma and melanoma were described over 30 years ago, and appear to explain at least some of the C3 deposited onto tumours [37–46].
Expression of CR2 by B cell lines is known to be required for activation of the alternative pathway [27,28], and the absence of CR2 on 721.221 cells prevented their use as class I− targets in this study. CR1 expressed by the normal B cell-derived line had somewhat of an opposite effect. CR1 both inhibited C3 deposition and promoted the breakdown of fixed iC3b into fixed C3dg. Although CR3 has a reduced affinity for C3dg compared with iC3b, experiments with β-glucan-primed NK cells have shown significant CR3-dependent cytotoxicity of targets bearing exclusively C3dg or C3d (V. Vetvicka and G. D. Ross, unpublished observation). This is potentially important, because some melanoma tumours have been shown to express cathepsin L that is capable of cleaving surface-bound iC3b into C3d [47,48].
As expected, unactivated NK cells failed to promote significant cytotoxicity in a 4-h assay with either B cell line, even when targets were opsonized with iC3b/C3dg. Although B cell lines are frequently used as targets for NK cell-mediated cytotoxicity, such assays require either IL-2 to activate NK cells or a 12–15-h assay period to allow for spontaneous NK cell activation and killing [49]. As previously shown with breast tumour cells [11], β-glucan failed to induce cytotoxicity of B cell lines lacking the CR3 target ligand iC3b. Only unopsonized K562 cells exhibit sensitivity to β-glucan-primed CR3-dependent NK cell cytotoxicity, presumably because K562 cells express a ligand for CR3 that is yet to be identified [10,50].
The reduced sensitivity of the class I+ B cell line to autologous NK cells was reversed by an IgG1 MoAb to class I (DX17) that is known to block the recognition domains of both the KIR and CD94/NKG2 [33,34]. An IgG1 MoAb to class II had no effect on NK cell cytotoxicity of either target cell. This confirmed the role of autologous class I in suppressing NK cell CR3-dependent cytotoxicity, presumably through KIR and/or CD94/NKG2 recognition.
Data gathered by many laboratories about the NK cell recognition system for class I have suggested that this may represent a barrier that protects class I+ tumours from destruction by NK cells. Although the presence of class I potentially allows tumours to be recognized by cytotoxic T lymphocytes (CTL) and provides the basis for development of vaccines based on tumour antigen peptides designed to fit class I binding grooves, recent reports have suggested that the selective pressure of CTL may be the driving force in the loss of class I expression by metastatic tumours [51,52]. Thus, it has been proposed that immunotherapy of metastatic disease should be based on NK cells, as the loss of class I by metastatic tumour cells makes a CTL-based immunotherapy useless while at the same time opening the door to NK cells by removing the KIR/CD94/NKG2 barrier [29]. Adoptive therapy with IL-2-activated lymphocytes does not result in the specific targeting of NK cells to tumours and is associated with significant toxicity [29,53–55]. By contrast, soluble β-glucan has the potential of targeting NK cells (as well as CR3-bearing macrophages and neutrophils) to tumour cells bearing fixed iC3b. The presence of iC3b on tumours appears to be a common occurrence, and could be augmented with vaccines that elicit a humoral antibody response and do not require tumour cell expression of class I or class II. Moreover, soluble β-glucan therapy has been shown to be associated with little toxicity or side-effects [56].
Acknowledgments
The authors are grateful to several colleagues for the generous donation of MoAbs: John Atkinson, Washington University; Václav Horejsí, Czech Academy of Sciences; Peter Lachmann, Cambridge University; Lewis Lanier, DNAX Research Institute; Jerome Ritz, Harvard University. This work was supported by a grant from the National Institutes of Health, United States Public Health Service: R01 AI-27771-17.
References
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Hogg N, Berlin C. Structure and function of adhesion receptors in leukocyte trafficking. Immunol Today. 1995;16:327–30. doi: 10.1016/0167-5699(95)80147-2. [DOI] [PubMed] [Google Scholar]
- 3.Sugimori T, Griffith DL, Arnaout MA. Emerging paradigms of integrin ligand binding and activation. Kidney Int. 1997;51:1454–62. doi: 10.1038/ki.1997.199. [DOI] [PubMed] [Google Scholar]
- 4.Petty HR, Todd RF. Receptor–receptor interactions of complement receptor type 3 in neutrophil membranes. J Leuk Biol. 1993;54:492–4. doi: 10.1002/jlb.54.5.492. [DOI] [PubMed] [Google Scholar]
- 5.Ross GD, Vetvicka V. CR3 (CD11b,CD18): a phagocyte and NK cell membrane receptor with multiple ligand specificities and functions. Clin Exp Immunol. 1993;92:181–4. doi: 10.1111/j.1365-2249.1993.tb03377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sutterwala FS, Rosenthal LA, Mosser DM. Cooperation between CR1 (CD35) and CR3 (CD11b/CD18) in the binding of complement-opsonized particles. J Leukocyte Biol. 1996;59:883–90. doi: 10.1002/jlb.59.6.883. [DOI] [PubMed] [Google Scholar]
- 7.Diamond MS, Garcia-Aguilar J, Bickford JK, Corbi AL, Springer TA. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J Cell Biol. 1993;120:1031–43. doi: 10.1083/jcb.120.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cain JA, Newman SL, Ross GD. Role of complement receptor type three and serum opsonins in the neutrophil response to yeast. Complement Inflamm. 1987;4:75–86. doi: 10.1159/000463011. [DOI] [PubMed] [Google Scholar]
- 9.Thornton BP, Vetvicka V, Pitman M, Goldman RC, Ross GD. Analysis of the sugar specificity and molecular location of the β-glucan-binding lectin site of complement receptor type 3 (CD11b/CD18) J Immunol. 1996;156:1235–46. [PubMed] [Google Scholar]
- 10.Vetvicka V, Thornton BP, Ross GD. Soluble β-glucan polysaccharide binding to the lectin site of neutrophil or NK cell complement receptor type 3 (CD11b/CD18) generates a primed state of the receptor capable of mediating cytotoxicity of iC3b-opsonized target cells. J Clin Invest. 1996;98:50–61. doi: 10.1172/JCI118777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vetvicka V, Thornton BP, Wieman TJ, Ross GD. Targeting of NK cells to mammary carcinoma via naturally occurring tumor cell-bound iC3b and β-glucan-primed CR3 (CD11b/CD18) J Immunol. 1997;159:599–605. [PubMed] [Google Scholar]
- 12.Moretta L, Mingari MC, Pende D, Bottino C, Biassoni R, Moretta A. The molecular basis of natural killer (NK) cell recognition and function. J Clin Immunol. 1996;16:243–53. doi: 10.1007/BF01541388. [DOI] [PubMed] [Google Scholar]
- 13.Yokoyama WM, Reichlin A, Margulies DH, Smith HR. MHC class I-dependent and -independent NK cell specificity. Chem Immunol. 1996;64:1–18. [PubMed] [Google Scholar]
- 14.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–93. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 15.Litwin V, Gumperz J, Parham P, Phillips JH, Lanier LL. Specificity of HLA class I antigen recognition by human NK clones: evidence for clonal heterogeneity, protection by self and non-self alleles, and influence of the target cell type. J Exp Med. 1993;178:1321–36. doi: 10.1084/jem.178.4.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Binstadt BA, Brumbaugh KM, Dick CJ, et al. Sequential involvement of Lck and SHP-1 with MHC-recognizing receptors on NK cells inhibits FcR-initiated tyrosine kinase activation. Immunity. 1996;5:629–38. doi: 10.1016/s1074-7613(00)80276-9. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu Y, DeMars R. Production of human cells expressing individual transferred HLA-A,-B,-C genes using an HLA-A,-B,-C null human cell line. J Immunol. 1989;142:3320–8. [PubMed] [Google Scholar]
- 18.Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature. 1985;314:537–9. doi: 10.1038/314537a0. [DOI] [PubMed] [Google Scholar]
- 19.O'Rear LD, Ross GD. Assays for membrane complement receptors. In: Shevach EM, Colligan J, editors. Current protocols in immunology. New York: John Wiley & Sons; 1994. pp. 13.4.1–13.4.18. [DOI] [PubMed] [Google Scholar]
- 20.Phillips JH, Gumperz JE, Parham P, Lanier LL. Superantigen-dependent, cell-mediated cytotoxicity inhibited by MHC class I receptors on T lymphocytes. Science. 1995;268:403–5. doi: 10.1126/science.7716542. [DOI] [PubMed] [Google Scholar]
- 21.Myones BL, Dalzell JG, Hogg N, Ross GD. Neutrophil and monocyte cell surface p150,95 has iC3b-receptor (CR4) activity resembling CR3. J Clin Invest. 1988;82:640–51. doi: 10.1172/JCI113643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bauume DM, Robertson MJ, Levine H, Manley TJ, Schow PW, Ritz J. Differential responses to interleukin 2 define functionally distinct subsets of human natural killer cells. Eur J Immunol. 1992;22:1–6. doi: 10.1002/eji.1830220102. [DOI] [PubMed] [Google Scholar]
- 23.Bryant J, Day R, Whiteside TL, Herberman RB. Calculation of lytic units for the expression of cell-mediated cytotoxicity. J Immunol Methods. 1992;146:91–103. doi: 10.1016/0022-1759(92)90052-u. [DOI] [PubMed] [Google Scholar]
- 24.Theofilopoulos AN, Perrin LH. Binding of components of the properdin system to cultured human lymphoblastoid cells and B lymphocytes. J Exp Med. 1976;143:271–89. doi: 10.1084/jem.143.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yefenof E, Klein G, Kvarnung K. Relationships between complement activation, complement binding, and EBV absorption by human hematopoietic cell lines. Cell Immunol. 1977;31:225–33. [Google Scholar]
- 26.Caudwell V, Porteu F, Calender A, Pangburn MK, Halbwachs-Mecarelli L. Complement alternative pathway activation and control on membranes of human lymphoid B cell lines. Eur J Immunol. 1990;20:2643–50. doi: 10.1002/eji.1830201218. [DOI] [PubMed] [Google Scholar]
- 27.Mold C, Nemerow GR, Bradt BM, Cooper NR. CR2 is a complement activator and the covalent binding site for C3 during alternative pathway activation by Raji cells. J Immunol. 1988;140:1923–9. [PubMed] [Google Scholar]
- 28.Schwendinger MG, Spruth M, Schoch J, Dierich MP, Prodinger WM. A novel mechanism of alternative pathway complement activation accounts for the deposition of C3 fragments on CR2-expressing homologous cells. J Immunol. 1997;158:5455–63. [PubMed] [Google Scholar]
- 29.Porgador A, Mandelboim O, Restifo NP, Strominger JL. Natural killer cell lines kill autologous β2-microglobulin-deficient melanoma cells: implications for cancer immunotherapy. Proc Natl Acad Sci USA. 1997;94:13140–5. doi: 10.1073/pnas.94.24.13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramos OF, Sármay G, Klein E, Yefenof E, Gergely J. Complement-dependent cellular cytotoxicity: lymphoblastoid lines that activate complement component 3 (C3) and express C3 receptors have increased sensitivity to lymphocyte-mediated lysis in the presence of fresh human serum. Proc Natl Acad Sci USA. 1985;82:5470–4. doi: 10.1073/pnas.82.16.5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambris JD, Dobson NJ, Ross GD. Release of endogenous C3b inactivator from lymphocytes in response to triggering membrane receptors for β1H globulin. J Exp Med. 1980;152:1625–44. doi: 10.1084/jem.152.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross GD, Lambris JD, Cain JA, Newman SL. Generation of three different fragments of bound C3 with purified factor I or serum. I. Requirements for factor H vs CR1 cofactor activity. J Immunol. 1982;129:2051–60. [PubMed] [Google Scholar]
- 33.D'Andrea A, Chang C, Phillips JH, Lanier LL. Regulation of T cell lymphokine production by killer cell inhibitory receptor recognition of self HLA class I alleles. J Exp Med. 1996;184:789–94. doi: 10.1084/jem.184.2.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazetic S, Chang C, Houchins JP, Lanier LL, Phillips JH. Human natural killer cell receptors involved in MHC class I recognition are disulfide-linked heterodimers of CD94 and NKG2 subunits. J Immunol. 1996;157:4741–5. [PubMed] [Google Scholar]
- 35.Schreiber RD, Pangburn MK, Medicus RG, Müller-Eberhard HJ. Raji cell injury and subsequent lysis by the purified cytolytic alternative pathway of human complement. Clin Immunol Immunopathol. 1980;15:384–96. doi: 10.1016/0090-1229(80)90050-1. [DOI] [PubMed] [Google Scholar]
- 36.Klein E, Di Renzo L, Yefenof E. Contribution of CR3, CD11b/CD18 to cytolysis by human NK cells. Mol Immunol. 1990;27:1343–7. doi: 10.1016/0161-5890(90)90041-w. [DOI] [PubMed] [Google Scholar]
- 37.Morton DL, Malmgren RA, Holme EC, Ketcham AS. Demonstration of antibodies against human malignant melanoma by immunofluorescence. Surgery. 1968;64:233–40. [PubMed] [Google Scholar]
- 38.Lewis MG, Ikonopisov RL, Nairn RC, Phillips TM, Fairley GH, Bodenham DC, Alexander P. Tumor-specific antibodies in human malignant melanoma and their relationship to extent of the disease. Br Med J. 1969;3:547–52. doi: 10.1136/bmj.3.5670.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irie K, Irie RF, Morton DL. Evidence for in vivo reaction of antibody and complement to surface antigens of human cancer cells. Science. 1974;186:454–6. doi: 10.1126/science.186.4162.454. [DOI] [PubMed] [Google Scholar]
- 40.McCoy JP, Hofheinz DE, Ng AB, Nordqvist S, Haines HG. Tumor-bound immunoglobulin in human gynecologic cancers. JNCI. 1979;63:279–83. [PubMed] [Google Scholar]
- 41.Crawford LV, Pim DC, Bulbrook RD. Detection of antibodies against the cellular protein p53 in sera from patients with breast cancer. Int J Cancer. 1982;30:403–8. doi: 10.1002/ijc.2910300404. [DOI] [PubMed] [Google Scholar]
- 42.Pfreundschuh M, Rohrich M, Piotrowski W, Berlit P, Penzholz H. Natural antibodies to cell-surface antigens of human astrocytoma. Int J Cancer. 1982;29:517–21. doi: 10.1002/ijc.2910290506. [DOI] [PubMed] [Google Scholar]
- 43.Lloyd KO, Old LJ. Human monoclonal antibodies to glycolipids and other carbohydrate antigens: dissection of the humoral immune response in cancer patients. Cancer Res. 1989;49:3445–51. [PubMed] [Google Scholar]
- 44.Niculescu F, Rus HG, Retegan M, Vlaicu R. Persistent complement activation on tumor cells in breast cancer. Am J Pathol. 1992;140:1039–43. [PMC free article] [PubMed] [Google Scholar]
- 45.Disis ML, Calenoff E, McLaughlin G, et al. Existent T-cell and antibody immunity to HER-2/neu protein in patients with breast cancer. Cancer Res. 1994;54:16–20. [PubMed] [Google Scholar]
- 46.Merimsky O, Shoenfeld Y, Chaitchik S, Yecheskel G, Fishman P. Antigens and antibodies in malignant melanoma. Tumor Biol. 1994;15:188–202. doi: 10.1159/000217892. [DOI] [PubMed] [Google Scholar]
- 47.Ollert MW, Frade R, Fiandino A, et al. C3-cleaving membrane proteinase. A new complement regulatory protein of human melanoma cells. J Immunol. 1990;144:3862–7. [PubMed] [Google Scholar]
- 48.Jean D, Hermann J, Rodrigues-Lima F, Barel M, Balbo M, Frade R. Identification on melanoma cells of p39, a cysteine proteinase that cleaves C3, the third component of complement: amino-acid sequence identities with procathepsin L. Biochem J. 1995;312:961–9. doi: 10.1042/bj3120961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Öhlén C, Bejarano MT, Grönberg A, et al. Studies of sublines selected for loss of HLA expression from an EBV-transformed lymphoblastoid cell line. Changes in sensitivity to cytotoxic T cells activated by allostimulation and natural killer cells activated by IFN or IL-2. J Immunol. 1989;142:3336–41. [PubMed] [Google Scholar]
- 50.Di Renzo L, Yefenof E, Klein E. The function of human NK cells is enhanced by β-glucan, a ligand of CR3 (CD11b/CD18) Eur J Immunol. 1991;21:1755–8. doi: 10.1002/eji.1830210726. [DOI] [PubMed] [Google Scholar]
- 51.Cordon-Cardo C, Fuks Z, Drobnjak M, Moreno C, Eisenbach L, Feldman M. Expression of HLA-A,B,C antigens on primary and metastatic tumor cell populations of human carcinomas. Cancer Res. 1991;51:6372–80. [PubMed] [Google Scholar]
- 52.Garrido F, Cabrera T, Concha A, Glew S, Ruiz-Cabello F, Stern PL. Natural history of HLA expression during tumour development. Immunol Today. 1993;14:491–9. doi: 10.1016/0167-5699(93)90264-L. [DOI] [PubMed] [Google Scholar]
- 53.Bubeník J. Tumor inhibitory effects of regional interleukin-2 administration and the role of lymphokine-activated killer cells. In: Lotzová E, Herberman RB, editors. Interleukin-2 and killer cells in cancer. 1. Boca Raton: CRC Press; 1990. pp. 235–44. [Google Scholar]
- 54.Thijs LG, Hack CE, Van Schijndel RJMS, et al. Activation of the complement system during immunotherapy with recombinant IL-2: relation to the development of side effects. J Immunol. 1990;144:2419–24. [PubMed] [Google Scholar]
- 55.Baars JW, Hack CE, Wagstaff J, et al. The activation of polymorphonuclear neutrophils and the complement system during immunotherapy with recombinant interleukin-2. Br J Cancer. 1992;65:96–101. doi: 10.1038/bjc.1992.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams DL, Sherwood ER, Browder IW, McNamee RB, Jones EL, Di Luzio NR. Pre-clinical safety evaluation of soluble glucan. Int J Immunopharmacol. 1988;10:405–14. doi: 10.1016/0192-0561(88)90127-0. [DOI] [PubMed] [Google Scholar]