

Abstract
NOD mice spontaneously develop autoimmune diabetes. One of the manipulations that prevent diabetes in NOD mice is infection with mycobacteria or immunization of mice with mycobacteria-containing adjuvant. Infection of NOD mice with Mycobacterium avium, done before the mice show overt diabetes, results in permanent protection of the animals from diabetes and this protective effect is associated with increased numbers of CD4+ T cells and B220+ B cells. Here, we investigate whether the M. avium-induced protection of NOD mice from diabetes was associated with changes in the expression of Fas (CD95) and FasL by immune cells, as well as alterations in cytotoxic activity, interferon-gamma (IFN-γ) and IL-4 production and activation of T cells of infected animals. Our data indicate that protection of NOD mice from diabetes is a Th1-type response that is mediated by up-regulation of the Fas–FasL pathway and involves an increase in the cytotoxicity of T cells. These changes are consistent with induction by the infection of regulatory T cells with the ability of triggering deletion or anergy of peripheral self-reactive lymphocytes that cause the autoimmune disease of NOD mice.
Keywords: diabetes, tolerance, apoptosis, cytotoxicity, Th1/Th2
INTRODUCTION
The NOD mouse is a classical animal model of spontaneous T cell-mediated autoimmune IDDM. Numerous reports have shown that the autoimmune process in NOD mice can be delayed or prevented by either antigen-specific or broader T cell-specific manipulations if they are performed early in the ontogeny of the immune system of the mice, i.e. before extensive insulitis has occurred [1–6].
Several mechanisms may account for induction of T cell tolerance to self antigens, namely deletion or anergy of autoreactive T cells, suppression/regulation of autoreactive lymphocytes by other T cells, or competitive antigen (or autoantigen) presentation to T lymphocytes [7–10]. The relative contributions of these mechanisms to the maintenance of self tolerance in vivo are yet to be defined. Deletion of peripheral T lymphocytes may be the result of apoptosis through activation of the Fas (CD95)–FasL pathway. Fas function(s) is important in the development and maintenance of the immune system. Several critical parameters of both humoral and cellular responses are controlled by Fas after initiation of an immune response. Fas-mediated apoptosis may be essential in controlling the expansion of activated/primed T cell clones. A regulatory role requiring Fas up-regulation on activated lymphocytes is to suppress unintended activation of autoreactive clones, thereby protecting against initiation of an autoimmune response; therefore, activation-induced cell death by Fas–FasL pathway may restore peripheral tolerance [11–16].
One of the manipulations that prevents diabetes in NOD mice is infection with mycobacteria (e.g. bacille Calmette–Guerin (BCG) bacilli) or immunization of mice with mycobacteria-containing adjuvant (Freund's complete adjuvant (FCA)) [17–19]. Previous work from this laboratory has shown that infection of NOD mice with Mycobacterium avium, done before the mice show overt diabetes, also results in permanent protection of the animals from diabetes [20]; this protective effect was associated with increased numbers of CD4+ T cells and B220+ B cells [21]. In this work we have investigated whether the M. avium-induced protection of NOD mice from diabetes was associated with changes in the expression of Fas and FasL by immune cells, as well as alterations in cytotoxic activity, interferon-gamma (IFN-γ) and IL-4 production and activation of T cells of infected animals. Our data indicate that protection of NOD mice from diabetes is a Th1-type response that is mediated by up-regulation of the Fas–FasL pathway and involves an increase in the cytotoxicity of T cells. These changes are consistent with induction by the infection of regulatory T cells with the ability of triggering deletion or anergy of peripheral self-reactive lymphocytes that cause the autoimmune disease of NOD mice.
MATERIALS AND METHODS
Mice
Breeding nuclei of NOD/Lt and NON/Lt mice were established in this research centre from animals purchased from the Jackson Laboratory (Bar Harbour, ME). NOD mice spontaneously develop type 1 diabetes, whereas NON mice are a related diabetes-resistant strain. Female NOD and NON mice were infected at 8 weeks of age, i.e. before overt diabetes was observed in NOD animals. All mice were confirmed to be negative for glucose in urine at this age. Colourimetric strips were used to monitor glycosuria (Combur-Test; Boehringer, Mannheim, Germany).
Mycobacterial infection
Mycobacterium avium strain ATCC 25291, serotype 2, was grown in liquid culture at 37°C in Middlebrook 7H9 broth (Difco Labs, Detroit, MI) containing 0.04% Tween-80. Mycobacterium avium is a mycobacterial species that causes human disease only in immune deficient individuals; a previous report from this laboratory showed that both NOD and NON mice are naturally resistant to this infectious agent [20]. The mycobacteria were harvested from liquid culture by centrifugation (6000 g) and washed three times in PBS, as described before [22–24]. The bacteria were suspended in saline containing 0.04% Tween-80 and diluted to a concentration of 2 × 108 viable bacilli of M. avium per ml. Eight-week-old NOD and NON mice were infected intraperitoneally with 0.5 ml of the M. avium suspension in saline (i.e. 108 viable bacilli per mouse). The animals were killed 1 month after infection. Age-matched control NOD and NON mice were inoculated with 0.04% Tween-80 in saline. Eight mice were used in each experimental group.
Monoclonal antibodies
MoAbs used in the flow cytometry analysis of splenic lymphocytes of the mice were the following: PE-labelled anti-CD4, PE-labelled anti-CD3, FITC-labelled anti-CD8 and FITC-labelled anti-CD4, FITC-labelled anti-IgM, PE-labelled anti-Fas, PE-labelled anti-CD69, and biotinylated-goat anti-rabbit, all purchased from PharMingen Inc. (San Diego, CA); FITC-labelled anti-IgG and PE-labelled streptavidin were purchased from Southern Biotechnology (Birmingham, AL) and purified rabbit anti-mouse FasL was purchased from Calbiochem (La Jolla, CA).
Spleen cell suspensions
A standard procedure was used to prepare exhaustive cell suspensions from spleen [25]. Viable cells were counted by a trypan blue exclusion test.
Flow cytometric analysis of cell surface markers of splenic cells
Splenic cells (106) were incubated with 50 μl of each MoAb preparation for 20 min on ice and in the dark. The dilutions of MoAbs were the following: PE–anti-CD4, 1:400; FITC–anti-CD4 and FITC–anti-CD8, all 1:200; FITC–anti-IgM, FITC–anti-IgG, PE–anti-Fas, PE–anti-CD69, purified FasL, biotin goat anti-rabbit and PE–avidin, all 1:100; and anti-CD3, 1:50. After the cell pellets were washed three times in PBS–NaN3 0.01%–fetal calf serum (FCS) 3%, their staining pattern was analysed using a Becton Dickinson FACSort flow cytometer interfaced to a Hewlett Packard computer. Dead cells and erythrocytes were excluded from the analysis using a combination of forward light scatter and propidium iodide (PI; Sigma Chemical Co., St Louis, MO) gating, as previously described [26].
Cytotoxicity assay
A non-radioactive assay was used to determine the cytotoxic potential of splenic lymphocytes obtained from infected and control mice, as described elsewhere [27]. Briefly, splenic lymphocytes were unspecifically stimulated overnight with concanavalin A (Con A) (Sigma). The blasts (effector (E) cells) originated this way were separated by a Percoll (Pharmacia, Uppsala, Sweden) gradient. Viable target (T) lymphocytes were stained with Calcein-AM (CAM; Molecular Probes, Eugene, OR) 100 nm (30 min incubation at 37°C). E and T cells were cultured together in RPMI 1640 medium (Gibco BRL Life Technologies Ltd) containing 10% FCS, 200 mml-glutamine, 10 mm HEPES (all from Gibco BRL) and 50 μm 2-mercaptoethanol (2-ME; Sigma), for 3 h (37°C, 5% CO2). Fifteen minutes before the end of the incubation, PI was added. After washing the cells three times in PBS, their staining pattern was analysed using a Becton Dickinson FACSort flow cytometer interfaced to a Hewlett Packard computer. The double stain with CAM and PI enabled the distinction of killed target cells. Determination of the percentage of specific cytotoxicity was done as previously described [27].
ELISPOT assay
Measurement of IFN-γ and IL-4 production by ELISPOT was performed as described elsewhere [28]. Before performance of the ELISPOT assay, 4 × 106 splenic lymphocytes were stimulated overnight, either with antigen (4 μg/ml M. avium sonicate) or with 3 μg/ml Con A (positive control), in RPMI 1640 medium containing 5% FCS (37°C, 5% CO2). The antibodies used for detection were anti-mouse IFN-γ, R46A2, anti-mouse IFN-γ–biotin, AN18, anti-mouse IL-4, 1D11, and anti-mouse IL-4–biotin, 24G2 (all kindly provided by Dr R. Appleberg, I.B.M.C., Porto, Portugal) and streptavidin–alkaline phosphatase (Jackson ImmunoResearch).
Statistical analysis
The numerical data were statistically compared using Student's t-test. We have considered two numerical populations to be significantly different if P < 0.05 or P < 0.01; these differences are, respectively, labelled with one or two asterisks in the figures.
RESULTS
To investigate the immune mechanisms underlying M. avium-induced prevention of diabetes in NOD mice, 8-week-old NOD and NON mice were intraperitoneally infected with the mycobacteria. After 1 month of infection, when major and stable changes of the host response to M. avium infection had already occurred [21], we characterized several immune parameters of the mycobacterial infected mice. The present experiments confirmed our previous report that M. avium infection causes in NOD mice (but not in NON animals) elevation in the numbers of CD4+ T cells and B220+ B cells [21]; we also confirmed a significant increase in the number of IgG+ B cells, both in NOD and NON mice. We have now investigated whether the protective effect from diabetes of the M. avium-infected mice was related to changes in the Fas–FasL pathway, the cytotoxic activity of T cells or production of IFN-γ and cell activation.
Expression of Fas and FasL
Flow cytometric analysis of splenic lymphocytes stained with either Fas or FasL MoAbs was performed. We observed that M. avium infection induced increased Fas expression in both CD4+ and CD8+ T cells. No significant changes in Fas expression were seen in IgM+ B cells; infection decreased Fas expression in IgG+ B cells (Fig. 1). Interestingly, we found that in control mice Fas expression is much lower in T cells of NOD mice than in T lymphocytes of NON animals (Fig. 2a), no difference being seen in Fas expression in B cells (Fig. 3). The expression of Fas in T lymphocytes of infected NOD mice reached levels similar to that observed on T cells of control NON mice (Fig. 2b). It has been documented that increase in Fas expression may not be enough for apoptosis induction of self-reactive cells; the phenomena may require FasL expression and interaction with Fas. Therefore, we also studied the expression of FasL in splenic T cells of the mice. As documented in Fig. 4, mycobacterial infection led to a significant enhancement in the number of FasL+ T lymphocytes, particularly of CD4+ T cells.
Fig. 1.
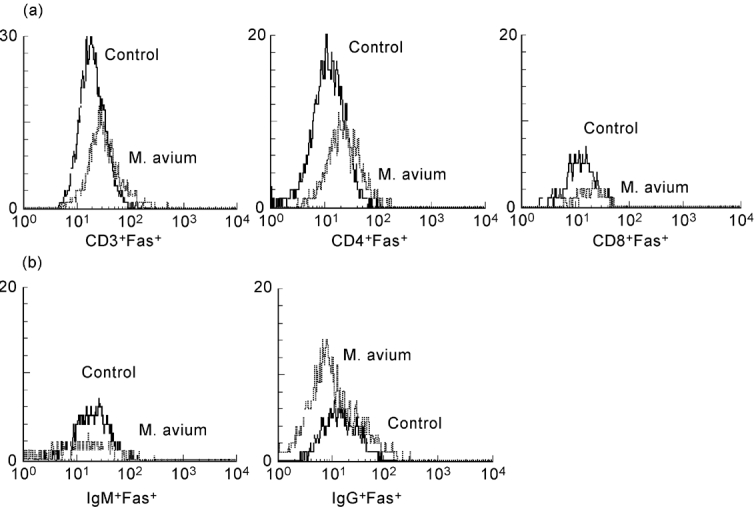
Mycobacterium avium infection induces an increase in Fas expression on T cells of NOD mice. Fas expression on the surface of T and B lymphocytes of NOD mice was measured by flow cytometry after mycobacterial infection or saline injection. There was a pronounced increase in Fas expression of T lymphocytes (a) after M. avium infection of NOD mice. Fas expression of B cells (b) remained similar to control levels of expression. n = 8.
Fig. 2.
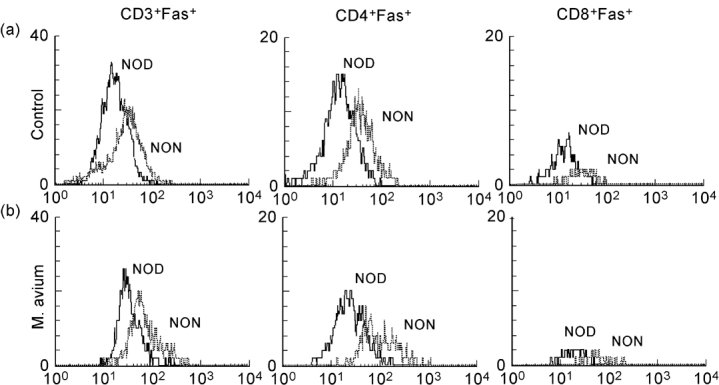
NOD mice T lymphocytes showed decreased Fas expression when compared with its related diabetes-resistant strain, NON mice. NOD and NON mice were intraperitoneally infected with 108Mycobacterium avium bacilli or injected with the vehicle of the bacteria. Fas expression on T lymphocytes was compared by flow cytometry. Control NOD mice showed deceased levels of Fas expression on T lymphocytes compared with control NON mice (a). Infection of NOD raised T lymphocyte Fas expression to levels similar to that observed in control NON mice (b). n = 8.
Fig. 3.
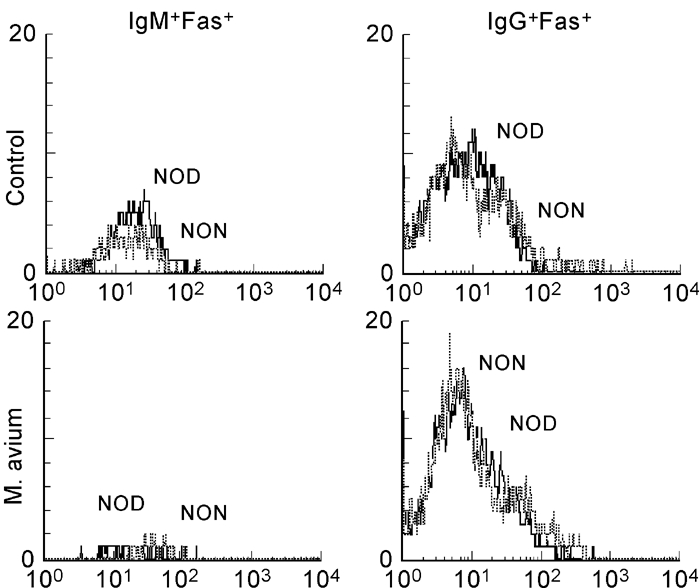
Fas expression on B lymphocytes is similar in NOD and NON mice. NOD and NON mice were intraperitoneally infected with 108Mycobacterium avium bacilli or injected with the vehicle of the bacteria. Fas expression on B lymphocytes was compared by flow cytometry. B lymphocytes from the two related strains showed similar levels of Fas. n = 8.
Fig. 4.
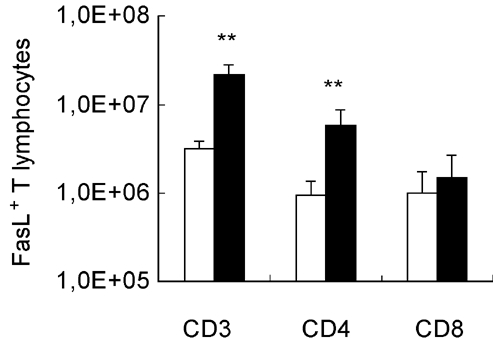
Mycobacteria-induced changes in the number of FasL+ T cells. FasL expression on the surface of T lymphocytes of NOD mice was measured by flow cytometry after mycobacterial infection or saline injection. The number of FasL+ cells was derived from two values: the percentage of FasL+ lymphocytes and the total number of splenic CD3+, CD4+ and CD8+ T cells. Mycobacterium avium infection induced a significant increase in the number of CD4+ T cells which express FasL. 1,0E + 08 = 108. n = 8. □, Control; ▪, M. avium.
Cytotoxicity of T cells
The cytotoxic potential of the activated T cells was assessed in vitro using lymphocytes as targets. We found that, in comparison with control mice, splenic lymphocytes from M. avium-infected NOD mice showed significantly increased cytotoxic activity against the target splenic cells (Fig. 5a). In contrast, infection did not result in an increased cytotoxic activity of T lymphocytes from NON mice (Fig. 5b).
Fig. 5.
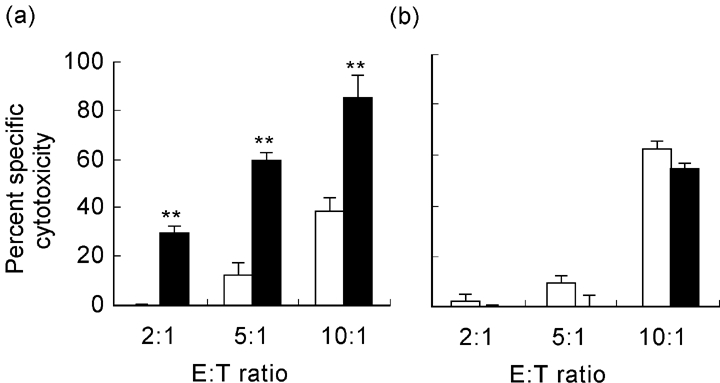
T lymphocytes of infected NOD mice showed increased cytotoxic potential. NOD and NON mice were intraperitoneally infected with 108Mycobacterium avium bacilli or injected with the vehicle of the bacteria. Lymphocytes obtained from either group of mice were used as effector cells in a cytotoxicity assay against splenic lymphocytes of untreated mice. T cells from infected NOD mice showed significantly increased cytotoxic potential (a); in contrast, there were no significant differences between cytotoxic activity of T cells from infected or control NON mice (b). Results refer to differences between groups of mice. n = 8. □, Control; ▪, M. avium.
IFN-γ and IL-4 production
An ELISPOT assay was performed to measure IFN-γ and IL-4 production by splenic lymphocytes of infected and control NOD mice. Splenic cells obtained from infected NOD mice showed increased IFN-γ production when compared with non-infected control mice. Interestingly, this effect was specific for the mycobacteria, as there were no differences in IFN-γ secretion between cells of infected and control mice when the cells were stimulated in vitro with Con A (Fig. 6a). IL-4 was detected in very small amounts in both groups of mice. Nevertheless, Con A-stimulated lymphocytes from control mice presented a significantly higher number of IL-4-producing cells than lymphocytes from infected mice (Fig. 6b).
Fig. 6.
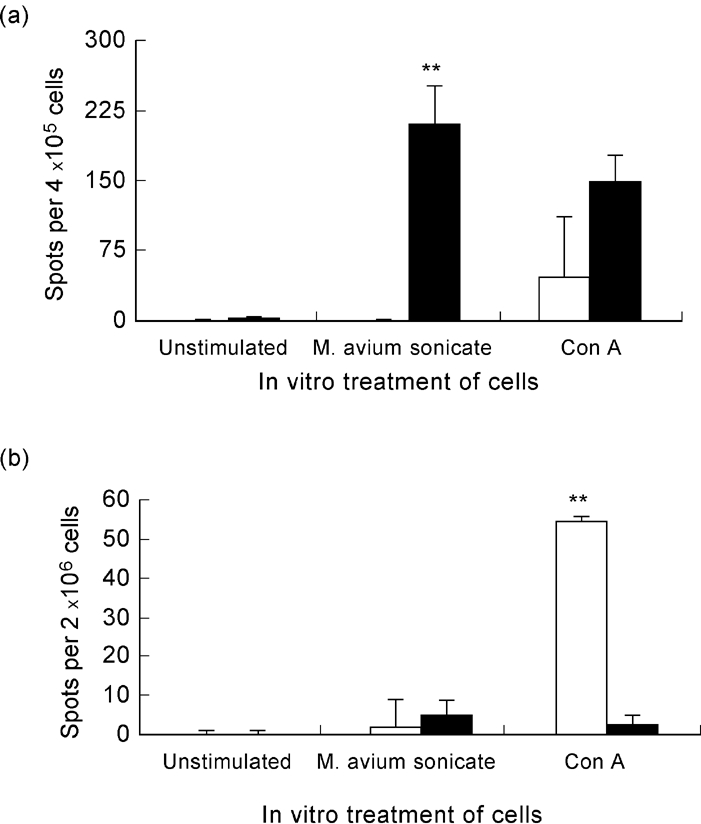
Response of NOD mice to mycobacteria includes IFN-γ production. IFN-γ and IL-4 production was measured by ELISPOT after in vitro stimulation of lymphocytes of either infected or control, saline-injected NOD mice, with sonicated mycobacteria. Concanavalin A (Con A) stimulation was used as a positive control. (a) There was a significant increase of IFN-γ-producing cells of infected mice after lymphocyte stimulation with sonicated Mycobacterium avium. No significant differences were observed between infected and control mice when Con A stimulation was performed. (b) There was an increase in the number of IL-4-producing cells of control mice after stimulation with Con A; in contrast, lymphocytes of infected mice showed almost no IL-4 production, whatever the in vitro stimulation performed. n = 8. □, Control; ▪, M. avium.
Expression of the early activation marker CD69
Mycobacterium avium infection of NOD mice induced a significant increase in the number of T cells (both CD4+ and CD8+) and B cells (both IgM+ and IgG+) expressing the CD69 surface antigen (Fig. 7). In contrast, infected NON mice only showed increased numbers of CD69+ IgG+ B cells and only a small increase in CD3+ T cells (that was not significant for CD4+ or CD8+ T cells) (Fig. 8).
Fig. 7.
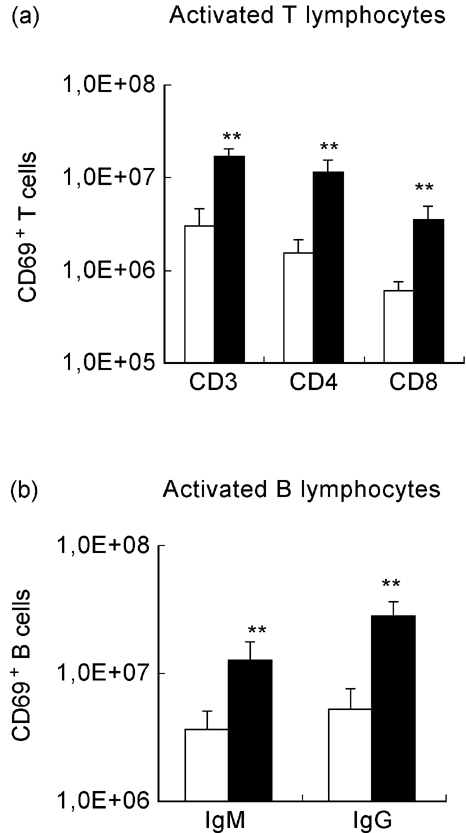
Activation state of lymphocytes from NOD mice after mycobacterial infection. NOD mice were intraperitoneally infected with 108 Mycobacterium avium bacilli or injected with the vehicle of the bacteria. Cell acquisition was performed gating each subset of lymphocytes (CD3+, CD4+, CD8+, IgM+, IgG+) and the percentage of CD69+ cells was determined. The number of CD69+ lymphocytes was derived from the percentage of CD69+ cells and the numbers of splenic CD3+, CD4+, CD8+, IgM+ and IgG+ lymphocytes. Mycobacterial infection induced significant increases in the numbers of activated, CD69-expressing T cells (both CD4+ and CD8+) and activated CD69+ B cells (expressing either surface IgM or IgG). 1,0E + 08 = 108. n = 8. □, Control; ▪, M. avium.
Fig. 8.
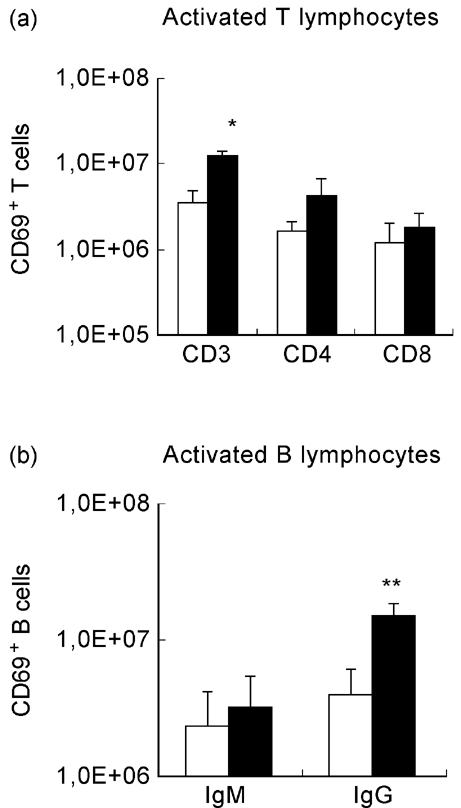
Activation state of lymphocytes from NON mice after mycobacterial infection. Cell acquisition was performed gating each subset of lymphocytes (CD3+, CD4+, CD8+, IgM+, IgG+) and the percentage of CD69+ cells was determined. The number of CD69+ lymphocytes was derived from the percentage of CD69+ cells and the numbers of splenic CD3+, CD4+, CD8+, IgM+ and IgG+ lymphocytes. NON mice were intraperitoneally infected with 108Mycobacterium avium bacilli or injected with the vehicle of the bacteria. M. avium infection induced a significant increase in the number of CD69-expressing IgG+ B cells, no significant differences being seen in either T cells or IgM+ B cells. 1,0E + 08 = 108. n = 8. □, Control; ▪, M. avium.
DISCUSSION
We have shown before that M. avium infection of NOD mice prevents the development of autoimmune diabetes and is associated with numerical changes in subpopulations of lymphocytes [7,8]. Interestingly, Baxter et al. reported that mycobacterial infection induces systemic lupus erythematosus (SLE)-like autoantibodies in NOD mice [29]. We report now that T cells of these diabetes-resistant NOD mice show enhanced expression of Fas and FasL molecules, increased cytotoxic activity, and augmented production of IFN-γ and decreased secretion of IL-4.
Clement & Stamenkovic [30] have recently shown that susceptibility of lymphocytes to Fas-mediated apoptosis is correlated to Fas expression on the cellular surface of those cells. Indeed, they showed that low/high levels of Fas expression correlate with reduced sensitivity to death, whereas an intermediate Fas expression sensitizes cells to apoptosis. Fas cytotoxic signals may therefore be dependent on a critical level of receptor surface expression. The importance of Fas in the regulation of mature T cells and the persistence of memory is accentuated by the involvement of Fas in the maintenance of peripheral tolerance and by its possible deregulation by a soluble form of Fas molecule in autoimmune disease [31,32]. This view is reinforced by the recent observations that activation-induced cell death is mediated by Fas–FasL interactions [11,13,31,32]. Interestingly, we found that expression of Fas on T lymphocytes of diabetes-prone NOD mice is much lower than the observed Fas expression on T cells of mice from NOD's related non-autoimmune strain, the NON mice. Moreover, we observed that upon M. avium infection there is an up-regulation of Fas on T lymphocytes from NOD mice to levels that are similar to the ones observed in non-infected NON mice. Thus, one may argue that the constitutively low expression of Fas on T lymphocytes of NOD mice may lead to deficient deletion of autoreactive cells upon activation and, consequently, to the development of autoimmune diabetes. The protective effect of M. avium infection may therefore be the result of raising Fas expression of lymphocytes to levels that sensitize autoreactive T cells to undergo programmed cell death. We also observed that the mycobacterial infection induced increased numbers of FasL-expressing T cells in NOD mice. These data may indicate that the protective effect of M. avium infection may be favoured by the induction of peripheral deletion of autoreactive T cells via Fas–FasL. Furthermore, these molecules, along with related ones (TNF/TNFR1) have already been implicated by other authors in IDDM β-cell death [33,34].
The augmented cytotoxicity observed in infected NOD mice is consistent with a Th1-dominated immune response and could involve both Th1 CD4+ T cells (through Fas–FasL pathway) and CD8+ T cells (through perforin/granzyme and/or Fas–FasL pathways). The importance of these two subpopulations of T lymphocytes in establishment and maintenance of peripheral tolerance through deletion of self-reactive cells is already very well established [7,30,35]. Consistent with a Th1-dominated response is our finding of increased IFN-γ and decreased IL-4 production by lymphocytes of infected mice. Other reports also describe a role for Th1 T cells in IDDM suppression: Akhtar et al. [36] documented the existence of Th1 β-cell-reactive T cell clones that have acquired a protective effector function at least in part by down-modulating immune responses; Han and co-workers [37] also identified CD4+, IFN-γ-producing, suppressor/regulatory T cells. They postulated that transforming growth factor-beta (TGF-β) also induced expansion of clones responsible for diabetes prevention, a hypothesis sustained by other investigations [38]. Finally, a beneficial role for IFN-γ in islet regeneration has also been postulated [7].
FasL expression has been correlated with the Th1 phenotype and it has been postulated that FasL+ Th1 (but not FasL+ Th2) cells are capable of lysing Th2, Th0 and Th1 cells by a Fas-mediated pathway [38]. Thus, Th1 lymphocytes may utilize Fas-mediated cell death to suppress Th2/Th0 as well as Th1 activity. β-cell destruction in IDDM is usually attributed to Th1-dominated response, whereas Th2 T cells are regarded as protective. However, there are other reports that disagree with the simple prediction that Th1 is pathogenic and Th2 is protective in diabetes of NOD mice. Wogensen and co-workers [39], for instance, have shown that transgenic expression of IL-10 in β-cells, which promotes a Th2-like cytokine pattern of islet-infiltrating cells, accelerated the onset of IDDM and led to an increased prevalence of the disease. Additionally, a recent work indicated that the inflammatory foci in NOD mice are dominated by a Th2 cytokine response [40]. Thus, β-cell destruction and T cell suppression of autoimmunity may involve a more complex interplay between Th1 and Th2 cells than is currently conceived. The Th1 CD4+ T cell subsets described here may be a part of the regulatory network that under normal circumstances plays a role in maintaining peripheral tolerance to self.
We propose that the protective effect of M. avium infection against diabetes of NOD mice may be achieved by peripheral deletion of autoreactive T lymphocytes via Fas–FasL. The event may result from molecular mimicry between mycobacterial antigen and pancreatic autoantigens, leading to deletion of lymphocytes reactive to β-cells as a consequence of the immune response directed to mycobacteria, or simply deletion of bystander autoreactive lymphocytes. The regulatory role of Fas up-regulation on activated lymphocytes could then be to suppress unintended activation of autoreactive clones, thereby protecting against the initiation of the autoimmune aggression of the pancreatic islets of NOD mice. In conclusion, our data suggest a mechanism to account for the protective effect of infection against the spontaneous autoimmune disease of NOD mice: infection induces deletion of peripheral autoreactive β-cell-specific T lymphocytes via Fas–FasL interactions effected by Th1-type T cells of enhanced cytotoxic activity.
Acknowledgments
The authors are grateful to Professor Carlos Martinez-A for having accepted one of us (T.C.M.) to work in his laboratory during a 6-month period. They also thank Dr Irene S. Leal (Institute for Molecular and Cell Biology, Porto, Portugal) for help in the performance of the ELISPOT assays. This work was supported by grants from EC and from JNICT (Portuguese Research Council).
References
- 1.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Exp Anim. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 2.Miller BJ, Appel MC, O'Neil JJ, Wicker LS. Both the Lyt2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J Immunol. 1988;140:52–8. [PubMed] [Google Scholar]
- 3.Hutchings PR, Simpson E, O'Reilly LA, Lund T, Waldmann H, Cooke A. The involvement of Ly2+ T cells in beta cell destruction. J Autoimmun. 1990;3:101–9. doi: 10.1016/s0896-8411(09)90018-x. [DOI] [PubMed] [Google Scholar]
- 4.Yagi H, Matsumoto M, Kunimoto K, Kawaguchi J, Makino S, Harada H. Analysis of the roles of CD4+ and CD8+ T cells in autoimmune diabetes of NOD mice using transfer to NOD athymic nude mice. Eur J Immunol. 1992;22:2387–93. doi: 10.1002/eji.1830220931. [DOI] [PubMed] [Google Scholar]
- 5.O'Garra A, Murphy K. T-cell subsets in autoimmunity. Curr Opin Immunol. 1993;5:880–6. doi: 10.1016/0952-7915(93)90100-7. [DOI] [PubMed] [Google Scholar]
- 6.Birk OS, Cohen IR. T-cell autoimmunity in type 1 diabetes mellitus. Curr Opin Immunol. 1993;5:903–9. doi: 10.1016/0952-7915(93)90104-z. [DOI] [PubMed] [Google Scholar]
- 7.Parish NM, Hutchings PR, O'Reilly L, Quartey-Papafio R, Healey D, Ozegbe P, Cooke A. Tolerance induction as a therapeutic strategy for the control of autoimmune endocrine disease in mouse models. Immunol Rev. 1995;144:269–300. doi: 10.1111/j.1600-065x.1995.tb00073.x. [DOI] [PubMed] [Google Scholar]
- 8.Rabinovitch A. Immunoregulatory and cytokine imbalances in the pathogenesis of IDDM. Therapeutic intervention by immunostimulation? Diabetes. 1994;43:613–21. doi: 10.2337/diab.43.5.613. [DOI] [PubMed] [Google Scholar]
- 9.Goodnow CC. Balancing immunity and tolerance: deleting and tuning lymphocyte repertoires. Proc Natl Acad Sci USA. 1996;93:2264–71. doi: 10.1073/pnas.93.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ridgway WM, Weiner HL, Fathman CG. Regulation of autoimmune responses. Curr Opin Immunol. 1994;6:946–55. doi: 10.1016/0952-7915(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 11.Singer GG, Carrera AC, Marshak-Rothstein A, Martinez-A C, Abbas AK. Apoptosis, Fas and systemic autoimmunity: the MRL- lpr/lpr model. Curr Opin Immunol. 1994;6:913–20. doi: 10.1016/0952-7915(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 12.Krammer PH, Dhein J, Walczak H, et al. The role of APO-1- mediated apoptosis in the immune system. Immunol Rev. 1994;142:175–91. doi: 10.1111/j.1600-065x.1994.tb00889.x. [DOI] [PubMed] [Google Scholar]
- 13.Gill BM, Nishikata H, Chan G, Delovitch TL, Ochi A. Fas antigen and sphingomyelin-ceramide turnover-mediated signalling: role in life and death of T lymphocytes. Immunol Rev. 1994;142:113–25. doi: 10.1111/j.1600-065x.1994.tb00885.x. [DOI] [PubMed] [Google Scholar]
- 14.Krammer PH, Behrmann I, Daniel P, Deihn J, Debatin K-M. Regulation of apoptosis in the immune system. Curr Opin Immunol. 1994;6:279–89. doi: 10.1016/0952-7915(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 15.Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 16.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–65. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 17.Qin HY, Sadelain MWJ, Hitchon C, Lauzon J, Singh B. Complete Freund's adjuvant-induced T cells prevent the development and adoptive transfer of diabetes in nonobese diabetic mice. J Immunol. 1993;150:2072–80. [PubMed] [Google Scholar]
- 18.Shehadeh NN, LaRosa F, Lafferty KJ. Altered cytokine activity in adjuvant inhibition of autoimmune diabetes. J Autoimmun. 1993;6:291–300. doi: 10.1006/jaut.1993.1025. [DOI] [PubMed] [Google Scholar]
- 19.Qin HY, Singh B. BCG vaccination prevents insulin-dependent diabetes mellitus (IDDM) in NOD mice after disease acceleration with cyclophosphamide. J Autoimmun. 1997;10:271–8. doi: 10.1006/jaut.1997.0136. [DOI] [PubMed] [Google Scholar]
- 20.Brás A, Águas AP. Diabetes-prone NOD mice are resistant to Mycobacterium avium and the infection prevents autoimmune disease. Immunol. 1996;89:20–25. doi: 10.1046/j.1365-2567.1996.d01-717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martins TC, Águas AP. Changes in B and T lymphocytes associated with mycobacteria-induced protection of NOD mice from diabetes. J Autoimmun. 1996;9:501–7. doi: 10.1006/jaut.1996.0067. [DOI] [PubMed] [Google Scholar]
- 22.Águas AP, Esaguy N, Sunkel CE, Silva MT. Crossreactivity and sequence homology between the 65-kDa mycobacterial heat shock protein and human lactoferrin, transferrin and DRβ subsets of MHC class II molecules. Infect Immun. 1990;58:1461–70. doi: 10.1128/iai.58.5.1461-1470.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brás A, Águas AP. Mycobacteria-induced autoantibody production is associated with susceptibility to infection but not host propensity to develop autoimmune disease. Clin Exp Immunol. 1995;100:75–80. doi: 10.1111/j.1365-2249.1995.tb03606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martins TC, Águas AP. Stress modulates inflammation triggered by mycobacteria in autoimmunity-prone and normal mice. Inflamm Res. 1995;44:393–9. doi: 10.1007/BF01797867. [DOI] [PubMed] [Google Scholar]
- 25.Rocha B, Larsson EL, Freitas AA. Effects of hydroxyurea on concanavalin A-induced T cell proliferation. Depletion of T cell growth factor reactive and producing T lymphocytes. Scand J Immunol. 1984;19:315–21. doi: 10.1111/j.1365-3083.1984.tb00936.x. [DOI] [PubMed] [Google Scholar]
- 26.Bandeira A, Mota-Santos T, Itohara S, Degermann S, Heusser C, Tonewaga S, Coutinho A. Localization of γ/δ T cells to the intestinal epithelium is independent of normal microbial colonization. J Exp Med. 1990;172:239–44. doi: 10.1084/jem.172.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papadopoulos NG, Dedoussis GVZ, Spanakos G, Gritzapis AD, Baxevanis CN, Papamichail M. An improved fluorescent assay for the determination of lymphocyte-mediated cytotoxicity using flow cytometry. J Immunol Methods. 1994;177:101–11. doi: 10.1016/0022-1759(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 28.Brandt L, Oettinger T, Holm A, Andersen AB, Andersen P. Key epitopes on the ESAT-6 antigen recognized in mice during the recall of protective immunity to Mycobacterium tuberculosis. J Immunol. 1996;157:3527–33. [PubMed] [Google Scholar]
- 29.Baxter AG, Horsfall AC, Healey D, Ozegbe P, Day S, Williams DG, Cooke A. Mycobacteria precipitate an SLE-like syndrome in diabetes-prone NOD mice. Immunology. 1994;83:227–31. [PMC free article] [PubMed] [Google Scholar]
- 30.Clement MV, Stamenkovic I. Fas and tumor necrosis factor receptor-mediated cell death: similarities and distinctions. J Exp Med. 1994;180:557–67. doi: 10.1084/jem.180.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng J, Zhou T, Liu C, Saphiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Sci. 1994;263:1759–62. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- 32.Singer GG, Abbas AK. The Fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1994;1:365–71. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 33.Benoist C, Mathis D. Cell death mediators in autoimmune diabetes—no shortage of suspects. Cell. 1997;89:1–3. doi: 10.1016/s0092-8674(00)80174-9. [DOI] [PubMed] [Google Scholar]
- 34.Chernovsky AV, Wang Y, Wong FS, Visintin I, Flavell RA, Janeway CA, Jr, Matis LA. The role of Fas in autoimmune diabetes. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, Inobe JI, Marks R, Gonnella P, Kuchroo VK, Weiner HL. Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature. 1995;376:177–80. doi: 10.1038/376177a0. [DOI] [PubMed] [Google Scholar]
- 36.Akhtar I, Gold JP, Pan LY, Ferrara JL, Yang XD, Kim JI, Tan KN. CD4+ beta islet cell-reactive T cell clones that suppress autoimmune diabetes in nonobese diabetic mice. J Exp Med. 1995;182:87–97. doi: 10.1084/jem.182.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han HS, Jun HS, Utsugi T, Yoon JW. A new type of CD4+ suppressor T cell completely prevents spontaneous autoimmune diabetes and recurrent diabetes in syngeneic islet-transplanted NOD mice. J Autoimmun. 1996;9:331–9. doi: 10.1006/jaut.1996.0045. [DOI] [PubMed] [Google Scholar]
- 38.Hahn S, Stalder T, Wernli M, Bürgin D, Tschopp J, Nagata S, Erb P. Down-modulation of CD4+ T helper type 2 and type 0 cells by T helper type 1 cells via Fas/Fas-ligand interaction. Eur J Immunol. 1995;25:2679–85. doi: 10.1002/eji.1830250942. [DOI] [PubMed] [Google Scholar]
- 39.Wogensen L, Lee MS, Sarvetnick N. Production of IL-10 by islet cells accelerates immune-mediated destruction of beta cells in non-obese diabetic mice. J Exp Med. 1994;179:1379–84. doi: 10.1084/jem.179.4.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson JT, Cornelius JG, Jarpe AJ, Winter WE, Peck AB. Insulin-dependent diabetes in the NOD mouse model. II. β cell destruction in autoimmune diabetes is a Th2 and not a Th1 mediated event. Autoimmun. 1993;15:113–22. doi: 10.3109/08916939309043886. [DOI] [PubMed] [Google Scholar]