

Abstract
The aim of the present study was to investigate the pathogenic properties of islet-infiltrating lymphocytes related to the severity of the autoimmune destruction of islet β-cells in the NOD mouse. We analysed the development of insulin-dependent diabetes mellitus (IDDM) produced by adoptive transfer of islet lymphocytes from NOD into NOD.scid mice. Here we show that the transfer was most effective when both CD4+ and CD8+ T cells were present in the infiltrate, but CD4+ T cells alone were sufficient to cause the disease. Islet lymphocytes from both females and males transferred diabetes effectively, but the severity of IDDM was higher when female islet lymphocytes were used. Unexpectedly, the sensitivity of male islets to β-cell damage was greater than that of female islets. Treatment of NOD females with a peptide of heat shock protein (hsp)60, p277, known to protect NOD mice from IDDM, reduced the pathogenicity of the islet lymphocytes. In contrast, administration of cyclophosphamide to males, a treatment that accelerates the disease, rendered the islet lymphocytes more pathogenic. More severe disease in the recipient NOD.scid mice was associated with more interferon-gamma (IFN-γ)-secreting islet T cells of the NOD donor. The disease induced by islet lymphocytes was strongly inhibited by co-transfer of spleen cells from prediabetic mice, emphasizing the regulatory role of peripheral lymphocytes. Thus, the cellular characteristics of the islet infiltrate and the pathogenicity of the cells are subject to complex regulation.
Keywords: insulitis, cytokines, adoptive transfer, therapy
INTRODUCTION
NOD mice spontaneously develop insulin-dependent diabetes mellitus (IDDM) due to the accumulation of mononuclear leucocytes in their pancreatic islets, insulitis, and the subsequent destruction of the insulin-secreting β-cells [1]. The NOD mouse disease shares many features with human type 1 diabetes mellitus, an autoimmune disease mediated by T cells: in both humans and NOD mice, antibody and T cell responses to several autoantigens are found, the disease may be transferred passively by T cells into a syngeneic recipient, and immunosuppression provides protection [2].
Although accumulating data provide strong evidence for the role of T cells in the pathogenesis of IDDM, very little is known about the properties of the lymphocytes that invade the islets. NOD mice of both sexes develop insulitis, but overt disease occurs mainly in females, and some colonies, like the NOD/Wehi [3], are characterized by a very low frequency of diabetes. Moreover, various immune manipulations have been reported to arrest the development of IDDM without eliminating the insulitis [4]. To investigate whether non-destructive insulitis might contain lymphocytes of lower pathogenicity, we developed an experimental system which allowed us to compare the pathogenic properties of islet lymphocytes directly. The system was based on the ability of islet-infiltrating T cells to transfer diabetes rapidly into NOD.scid mice. These mice have the NOD genes, but do not have mature T and B lymphocytes due to the scid mutation [5], and thus are free of IDDM, unless reconstituted with NOD lymphocytes. By injecting NOD.scid mice with equal numbers of NOD islet lymphocytes and monitoring the development of diabetes, we could show that the pathogenicity of insulitis is regulated by a variety of influences.
MATERIALS AND METHODS
Animals
Mice were bred at the Weizmann Institute animal breeding centre. NOD/Lt mice were housed in a specific pathogen-free environment. Normoglycaemic 2–4-month-old mice of both sexes were used as donors of islet and spleen cells. NOD.scid mice 2–3 months old were used as recipients.
Reagents
Tissue culture media RPMI 1640 and buffered salt solutions were obtained from the Weizmann Institute biological services. HEPES, fetal calf serum (FCS) and trypsin–EDTA solution were purchased from Biological Industries (Beit Haemek, Israel). Histopaque 1119, agarose, cyclophosphamide, 3,3′-diaminobenzidine (DAB), and 5-bromo-4-chloro-3-indolylphosphate (BCIP) were bought from Sigma (St Louis, MO). 2-amino-2-methyl-1-propanol (AMP) was obtained from Merck (Hohenbrunn, Germany), and collagenase P was purchased from Boehringer Mannheim (Mannheim, Germany). Streptavidin-alkaline phosphatase was obtained from Jackson ImmunoResearch Labs (West Grove, PA).
Matched pairs of rat anti-mouse cytokine MoAbs obtained from PharMingen (San Diego, CA) were: BVD4-1D11 (pure) and BVD6-24G2 (biotinylated) anti-IL-4, and R4-6 A2 (pure) and XMG1.2 (biotinylated) anti-interferon-gamma (IFN-γ). Anti-mouse CD3ε, CD4 and CD8a antibodies were monoclonal hamster 145-2C11, rat GK1.5 and rat 53–6.7, respectively (ATCC, Rockville, MD). The antibodies were purified from tissue culture supernatants by ammonium sulphate and dialysis, and were filter-sterilized. Immunostaining for insulin was performed using guinea pig anti-insulin and rabbit anti-guinea pig peroxidase-labelled antibodies purchased from Zymed Labs (South San Francisco, CA).
Peptide p277 of the human heat shock protein (hsp)60 [6], positions 437–460 (VLGGGVALLRVIPALDSLTPANED; with valines in place of cysteines at positions 6 and 11), and peptide MTp278 of Mycobacterium tuberculosis hsp60 molecule [7], positions 430–446 (EGDEATGANIVKVALEA) were prepared by Fmoc synthesis and purified by reverse phase high performance liquid chromatography (HPLC), as described [8].
Isolation of islet lymphocytes
Pancreatic islets were isolated by the intraductal collagenase technique [9], and purified by centrifugation on a density gradient of Histopaque (1.119-1.100-1.08). Islets were collected from the lower interface, pancreatic lymph nodes were removed by hand-picking, and the islets were treated with 0.25% trypsin–0.02% EDTA for 10 min at 37°C. The resulting single-cell suspension was washed, a sample centrifuged on slides, immunostained for insulin to confirm the islet origin of the epithelial cells, and the percentage of lymphocytes was counted. The islet cells were used either non-fractionated, or further processed to separate CD4+ or CD8+ T cells using the MiniMACS system (Miltenyi Biotec, Bergisch Gladbach, Germany) with magnetic microbeads coupled to the respective antibodies, according to the manufacturer's protocol. Typically, about 800 islets could be isolated from five prediabetic NOD mice at the age of 2–3 months, usually yielding 1–1.2 × 106 lymphocytes. Transfer experiments were not done unless there were sufficient donor cells to accommodate groups of no less than five recipient mice. From 60% to 80% of the islet lymphocytes were CD4+ T cells, judged by cell count before and after magnetic sorting or by immunohistochemical staining of non-fractionated cytospin samples.
Adoptive transfer of IDDM
Unless otherwise indicated, a mixed islet cell suspension corresponding to 2 × 105 lymphocytes was injected in 0.5 ml PBS intraperitoneally into NOD.scid mice. Since it was technically impossible to isolate islets from more than 10 mice at a time, no more than 10 recipient mice could be injected on the same day (two groups of 5). Therefore, the experiments with n > 5 represent data pooled from several experiments done on separate days. For co-transfer, spleen cells, 6 × 106/mouse, were injected intraperitoneally on the same day. For in vivo depletion, recipient mice were injected intraperitoneally with anti-CD4 or anti-CD8 antibodies at a dose of 200 μg/mouse, twice a week for 6 weeks. Blood glucose was monitored weekly by the ‘Companion 2’ blood glucose analyser (MediSense, Waltham, MA) with a working range of 1.1–33 mmol/l. Mice were considered diabetic if their blood glucose exceeded 13.5 mmol/l (250 mg/dl) on two consecutive determinations. Mean blood glucose values, cumulative incidence of the disease, and mortality rates were compared to assess relative IDDM severity. Mice were followed up to 12 weeks or until death.
Cytokine ELISPOT assay
The gel substrate method [10] was used. Briefly, sterile ELISA plates were precoated with anti-CD3 (5 μg/ml) and anti-cytokine capture antibodies (2 μg/ml) in PBS at 4°C, overnight. The plates were washed and islet or spleen cells were seeded in RPMI 1640–10% FCS, and incubated for 18 h at 37°C. Then the wells were washed with PBS, and biotinylated (detection) antibodies (4 μg/ml) were added for 2 h. This was followed by the addition of streptavidin-alkaline phosphatase (1:1000) and warm BCIP/AMP in 0.6% agarose as a substrate. Blue spots, each representing an individual cytokine-secreting cell (spot-forming cell (SFC)), were counted the next day. The results are expressed as the mean SFC ± s.e.m. of duplicate wells per indicated number of lymphoid cells.
Statistical analysis
The non-parametric unpaired Mann–Whitney test was used for comparison of the differences in mean blood glucose values. The frequencies of cytokine SFC were compared by Student's two-tailed unpaired t-test. The cumulative incidence of disease and the mortality were compared using Fisher's exact test. P < 0.05 was considered significant.
RESULTS
Transfer of islet-associated lymphocytes from NOD mice into MHC-matched NOD.scid recipients resulted in the rapid development of insulitis and β-cell loss (Fig. 1) with subsequent IDDM. As few as 105 islet lymphocytes were capable of rendering NOD.scid mice diabetic within 4 weeks. This high efficiency of disease transfer made it possible to analyse the T cell populations mediating β-cell destruction.
Fig. 1.
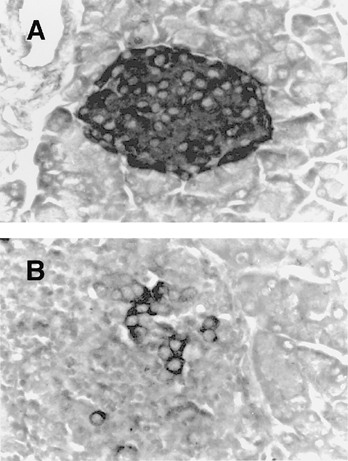
Immunohistochemical staining for insulin in NOD.scid islets. (A) An intact pancreatic islet of the NOD.scid mouse containing numerous insulin-positive cells (dark colour) and no mononuclear infiltration. (B) A typical destructive insulitis 4 weeks after the transfer of 2 × 105 NOD islet lymphocytes. Note the disappearance of the insulin-positive cells. (Immunoperoxidase-DAB, haematoxylin counterstain, × 400.)
Islet CD8+ T cells potentiate IDDM transfer by CD4+ cells
Figure 2a shows that negative selection of the donor population of either the CD4+ or the CD8+ T cells only slightly reduced the ability of the transferred lymphocytes to cause diabetes in the NOD.scid recipients. Although the mean blood glucose was significantly lower in the CD8-depleted group by week 8 after transfer, the cumulative incidence of IDDM (nine of 10) and the mortality (one of 10) by week 10 in this group was similar to that observed in the NOD.scid recipients that had been injected with non-depleted islet lymphocytes (10 of 10 and four of 10, respectively; NS). Similarly, depletion of the CD4+ T cells did not result in either a lower incidence (eight of 10), or a lower mortality (four of 10); NS. However, further depletion of CD4+ T cells by antibody treatment in vivo completely abrogated the development of IDDM (Fig. 2b). The incidence of IDDM in this group by week 6 was 0 of five compared with seven of 10 in the control group (P = 0.026). By week 12, all the mice in the anti-CD4-treated group were healthy, while in the control group all the mice were sick (P = 0.0003). By week 11, six of 10 mice in the control group were dead (P = 0.044). In contrast, administration of anti-CD8 antibody did not block the transfer of disease: three of five NOD.scid recipients became diabetic (NS compared with non-treated controls). However, the mean blood glucose in this group was lower than in the non-treated mice by weeks 6 and 7 (Fig. 2b) and none of the mice were dead by week 11 (0 of five compared with six of 10; P = 0.044). These results indicate that islet-infiltrating CD4+ and CD8+ T cells are both necessary for effective transfer of diabetes, but the CD4+ T cells are indispensable. The essential role of the CD4+ T cells was confirmed by transfer of pure CD4+ or CD8+ T cells obtained by positive selection: the CD4+ fraction was able to cause diabetes (Fig. 2c), while the CD8+ fraction was not. Therefore, CD4+ islet T cells are necessary and sufficient for β-cell destruction. Nevertheless, the transfer of IDDM was most effective when both CD4+ and CD8+ populations were present. We therefore used non-fractionated islet lymphocytes for further experiments.
Fig. 2.
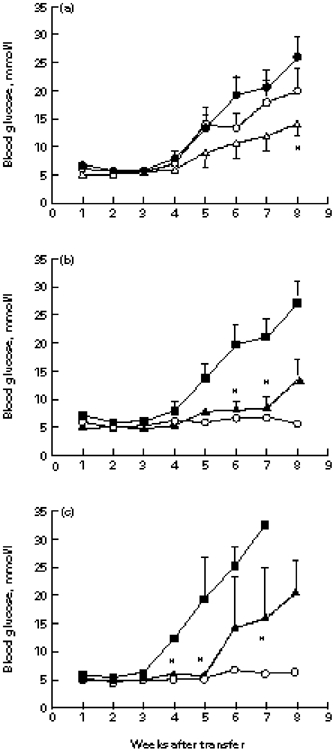
The effect of islet CD4+ and CD8+ T cells in the adoptive transfer of insulin-dependent diabetes mellitus (IDDM). (a) Negative selection of CD4+ or CD8+ T cells by magnetic sorting. Islet lymphocytes were incubated in vitro with MiniMACS anti-CD4 or anti-CD8 beads and separated on a magnetic column. Groups of 10 NOD.scid mice were injected intraperitoneally with equal cell numbers, and blood glucose was monitored. Data are shown as the mean ± s.e.m. *P < 0.05. •, Non-selected; ○, CD4-depleted; Δ, CD8-depleted. (b) Groups of 5–10 NOD. scid mice were injected with CD4- or CD8-depleted islet lymphocytes as in (a), and treated in vivo with either anti-CD4 or anti-CD8 MoAb. *P < 0.05 compared with the non-treated control group. ▪, No treatment; ▴, CD8/anti-CD8; ○, CD4/anti-CD4. (c) Adoptive IDDM transfer with positively selected islet CD4+, CD8+, or both CD4+ plus CD8+ T cells. In NOD.scid recipients of CD4+ T cells, the mean blood glucose was significantly lower than was observed in the recipients of both populations by weeks 4, 5 and 7 (*P < 0.05). ▪, CD4+ and CD8+; ▴, CD4+; ○, CD8+.
Gender-dependent differences in islet effector and target cells
To learn whether the lower incidence of spontaneous diabetes in NOD males might be due to a qualitative difference in their islet-infiltrating lymphocytes, we transferred equal numbers of lymphocytes from male or female NOD islets into female NOD.scid mice and monitored the development of diabetes. Table 1 shows that the male islet lymphocytes were capable of transferring diabetes at an incidence similar to that of the female cells. However, the severity of the disease induced in females by male cells was significantly lower by week 5 (Fig. 3a). Consequently, eight of 10 NOD.scid recipients that had received female islet lymphocytes were dead by week 6, while none of the five recipients of male cells died (P = 0.007).
Table 1.
Cumulative incidence of insulin-dependent diabetes mellitus transferred by islet lymphocytes
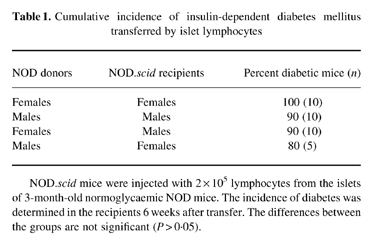
NOD.scid mice were injected with 2 × 105 lymphocytes from the islets of 3-month-old normoglycaemic NOD mice. The incidence of diabetes was determined in the recipients 6 weeks after transfer. The differences between the groups are not significant (P > 0.05).
Fig. 3.
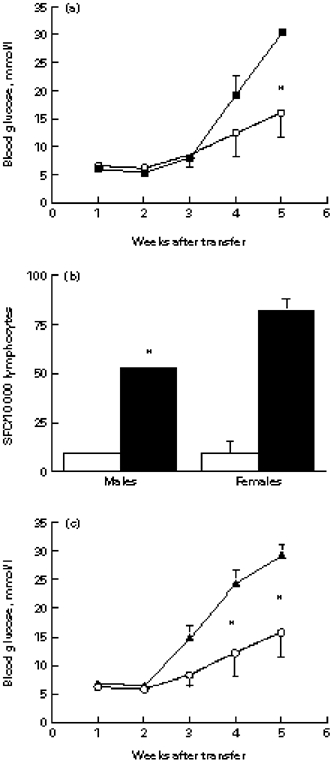
Gender differences in islet effector and target cells. (a) Islet lymphocytes from 3-month-old male or female NOD mice were injected into groups of 5–10 female NOD.scid recipients and the blood glucose was monitored. (*P < 0.025). ▪, Female to female; ○, male to female. (b) Frequencies of islet T cells capable of secreting IFN-γ (▪) and IL-4 (□). Male or female NOD islet T cells were activated by anti-CD3 MoAb and the numbers of cytokine producers counted as spot-forming cells (SFC). Male lymphocytes contained less IFN-γ producers than did the female lymphocytes (*P < 0.05). (c) Islet lymphocytes from 3-month-old NOD males were injected into female (n = 5) or male (n = 10) NOD.scid recipients and blood glucose was monitored (*P < 0.03). ○, Male to female; ▴, male to male.
Since the recipient mice were females, the anti-H-Y response and the influence of male hormones were excluded. We concluded that the difference in IDDM severity had to be caused by the intrinsic properties of the donor lymphocytes. We therefore analysed the frequencies of male and female NOD islet T cells capable of secreting the representative Th1 and Th2 cytokines, IFN-γ and IL-4, in response to CD3 cross-linking. Figure 3b shows that the frequencies of IFN-γ-secreting islet T cells in the females were 55% higher than in the males (82.6 ± 6.2 compared with 53.4 ± 0.2 per 104 lymphocytes, respectively; P < 0.05), while the frequencies of IL-4 producers were not different (9.5 ± 0.7 and 9.6 ± 1.6 per 104 lymphocytes, respectively).
It was conceivable that the higher incidence of IDDM in female NOD mice might also result from a greater sensitivity of their target β-cells to autoimmune damage. To test this possibility, we transferred male NOD islet cells into NOD.scid male and female mice. Contrary to expectations, the female recipients developed less severe hyperglycaemia than did the males (Fig. 3c), with lower mortality (0 of five compared with six of 10, respectively, at week 6; P = 0.044), suggesting that the β-cells of NOD females might actually be more resistant to damage than those of males. Thus, the difference between females and males in β-cell killing is more a function of the infiltrating effector cells than of the sensitivity of the target β-cells.
p277 peptide treatment renders islet lymphocytes less pathogenic
To learn whether the ability of islet-infiltrating lymphocytes to destroy β-cells could be inhibited by treatment, we used a model for protection from IDDM by administration of an hsp60 peptide, p277 [6,7]. Control mice were treated with an immunogenic peptide of M. tuberculosis hsp60, MTp278. Female NOD mice were treated with a single s.c. dose of 100 μg of either peptide in Freund's incomplete adjuvant (FIA) at the age of 3 months. Six weeks later, the islet lymphocytes were transferred into NOD.scid recipients. We found that the donors treated with p277 transferred IDDM at a slower rate (only two of eight mice were sick at week 7 after transfer, compared with eight of 10 in the control peptide; P = 0.023), and with delayed mortality (two of eight mice compared with nine of 10 mice in the control group at week 11; P = 0.0128). The mean blood glucose levels were significantly lower at week 10 post-transfer (Fig. 4a). The decreased pathogenicity of the islet-infiltrating lymphocytes from the p277-treated NOD mice was associated with a lower concentration of IFN-γ producers, compared with both the PBS- and MTp278-treated donors (Fig. 4b). There were no significant differences in the numbers of IL-4 producers between the p277- and MTp278-treated groups.
Fig. 4.
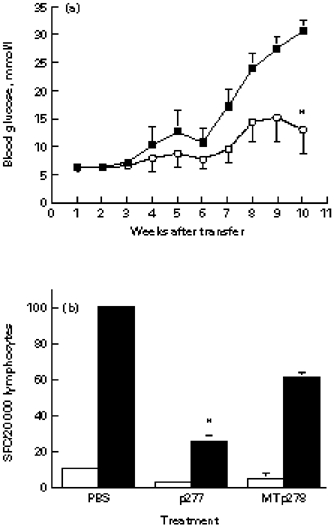
The effect of peptide treatment of NOD female mice on islet T cell pathogenicity and cytokine production. (a) Three-month-old NOD donor female mice were treated with a single s.c. injection of 100 μg of either p277 (○) or MTp278 peptide (▪) in Freund's incomplete adjuvant (FIA). Six weeks later, the donor islet lymphocytes (1.5 × 105 cells per mouse) were injected into NOD.scid females, and the development of diabetes was monitored. *P < 0.048; n = 8 for the p277 group; n = 10 for the MTp278 group. (b) The effect of p277 treatment on the frequency of islet T cells capable of secreting IFN-γ. Donor mice were treated as above (a), and their islet lymphocytes were analysed by the ELISPOT assay for numbers of IFN-γ (▪) and IL-4 producers (□) (SFC), that could be activated by anti-CD3. The differences between the IFN-γ SFC of the p277-treated group compared with the groups treated with MTp278 or PBS were significant (*P < 0.05).
Effects of cyclophosphamide
Diabetes may be accelerated in NOD mice by the administration of cyclophosphamide [11]. In order to test the effect of cyclophosphamide on the pathogenicity of islet lymphocytes, we treated NOD males with two doses of 200 mg/kg intraperitoneally and, 3 days after the last injection, isolated the islets for transfer. The islet lymphocytes from the cyclophosphamide-treated mice induced a more severe diabetes in the NOD.scid recipients than did the lymphocytes from the PBS-treated control mice (Fig. 5a), and contained more IFN-γ producers (Fig. 5b). It has been suggested that cyclophosphamide acts by inhibiting suppressor T cells [12]. If this is true, treatment of the recipient NOD.scid mice with cyclophosphamide after the transfer of NOD islet lymphocytes should also exacerbate the disease. However, no acceleration of IDDM occurred in the cyclophosphamide-treated group of recipients compared with the PBS-treated controls; actually, the development of IDDM was markedly inhibited (Fig. 5c). Cyclophosphamide-treated NOD.scid recipients developed IDDM at a lower incidence (0 of five compared with five of 10 at week 4; P = 0.002), and with a lower mortality rate (0 of five compared with seven of 10 at week 7; P = 0.026) compared with PBS-treated controls. This suggests that some pathogenic islet lymphocytes also may be sensitive to cyclophosphamide.
Fig. 5.
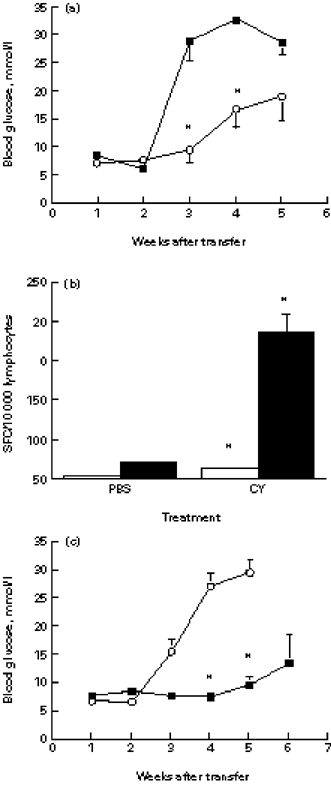
Effects of cyclophosphamide (CY) on islet T cell pathogenicity and cytokine production. (a) Two-month-old male NOD mice were treated with CY (▪) or with PBS (○), their islet lymphocytes were transferred into groups of 5–10 male NOD.scid mice, and blood glucose was monitored. *P < 0.015. (b) Islet lymphocytes were isolated from CY-treated or PBS-treated normoglycaemic NOD males and analysed for the frequencies of IFN-γ- (▪) and IL-4-secreting T cells (□) by the ELISPOT assay. The mean SFC of duplicate wells are shown from one of two similar experiments. *P < 0.05 compared with PBS-treated controls. (c) Male NOD.scid mice, five per group, were injected with male NOD islet lymphocytes and treated either with CY (▪) (n = 5) or PBS (○) (n = 10). The development of diabetes was monitored. *P < 0.003.
Splenocytes inhibit IDDM transfer
An impressive finding of the adoptive transfer experiments was the high incidence of the acquired disease (Table 1), suggesting that islet lymphocytes may be more aggressive in the NOD.scid environment than they are in the original NOD donor. The slower disease in the intact NOD mice might be due to protection provided by peripheral lymphocytes. To test this possibility, we co-transferred islet and spleen lymphocytes. Figure 6 shows that 2 × 105 islet lymphocytes rapidly transferred diabetes (nine of 10 mice were diabetic by week 4 after transfer), while none of the recipients of 6 × 106 NOD spleen cells became hyperglycaemic before week 10 (P = 0.002). Consequently, eight of 10 recipients of islet lymphocytes were dead by week 8 (P = 0.007). This difference could be explained merely by a higher frequency of specific effector cells in the insulitis than in the spleen. However, when the spleen cells were co-transferred with the islet cells, the development of IDDM was significantly inhibited. The incidence of IDDM was lower at week 3 (0 of five compared with six of 10 mice; P = 0.044), and at week 4 (0 of five compared with nine of 10 mice; P = 0.002). None of the mice transferred with the cell mixture died before week 11. Since the islet lymphocytes alone were sufficient to destroy the recipient β-cells, the inhibition provided by the addition of the spleen cells suggests that inhibitory cells were present in the spleen.
Fig. 6.
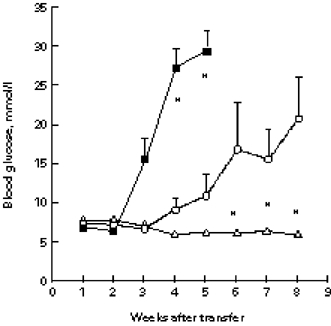
Inhibition of adoptively transferred insulin-dependent diabetes mellitus (IDDM) by spleen cells. Islet lymphocytes (▪) (2 × 105), spleen cells (Δ) (6 × 106), or a mixture of both (○) from male NOD mice were injected intraperitoneally into NOD.scid males, and blood glucose levels were measured weekly. *P < 0.02 compared with the group injected with the cell mixture.
DISCUSSION
During the three decades since insulitis was considered the major pathomorphological feature of IDDM [13], accumulating evidence indicates that leucocytic infiltration per se does not necessarily result in overt diabetes. Thus, destructive insulitis, associated with the β-cell loss, should be distinguished from non-destructive insulitis [14]. Indeed, it is possible to protect NOD mice from diabetes by various relatively mild strategies, which do not eliminate islet-infiltrating lymphocytes, but rather turn the destructive insulitis into a non-destructive insulitis [7,15,16]. However, are islet lymphocytes from mice with a low or a high incidence of IDDM qualitatively different?
We addressed this question using an adoptive transfer model, in which we injected freshly isolated islet leucocytes of NOD mice into NOD.scid recipients. This enabled us to compare the pathogenic potential of donor islet lymphocytes without any recruitment of recipient lymphocytes that might interact with the donor lymphocytes. Since equal numbers of lymphocytes were injected, any differences in the development of the transferred IDDM could be attributed to qualitative differences between the lymphocytes.
Our results demonstrate that IDDM transfer is most effective when both the CD4+ and CD8+ islet T cells are present, since depletion of either subset results in a significantly lower severity of disease (Fig. 2b). This observation is in agreement with previous reports of passive transfer using NOD spleen cells [17,18]. Hence, rapid β-cell destruction occurs when CD4+ and CD8+ islet-infiltrating T cells can co-operate. After negative selection in vitro of either subset, the islet lymphocytes were still able to transfer IDDM. However, we could not conclude that the CD4− and CD8− T cells can work independently, because depletion might have been incomplete. We therefore depleted the cell populations both in vitro and in vivo, and then found that the NOD.scid recipient mice treated with the anti-CD4 antibody did not develop IDDM during the entire 12-week observation period. In contrast, the mice treated with the anti-CD8 antibody did become sick, although the disease developed later and was less severe than in the non-treated group. Similarly, positively sorted CD4+ cells could transfer the disease, while CD8+ cells alone could not, indicating that the CD4+ T cells in the lesion are both necessary and sufficient. These results are in accord with data reported by others using islet-specific T cell clones [19–21], or in vitro plus in vivo depletion of splenic CD8+ cells [22], suggesting that CD4+ cells alone can destroy islet β-cells.
It was reported recently that the initiation of insulitis in NOD mice is MHC class I-restricted and therefore CD8+ T cell-dependent: spleen cells of prediabetic NOD mice could not transfer IDDM into β2-microglobulin-deficient NOD.scid mice [23]. Nevertheless, all the NOD donors in our experiments were prediabetic, but no absolute requirement for CD8+ cells was found, possibly because the lymphocytes were taken from the lesion, that is from an established insulitis. Lesional CD4+ T cells are already primed, and possibly might enter and destroy recipient islets without the aid of CD8+ T cells. However, CD8+ islet T cells clearly enhanced the pathogenicity of the CD4+ T cells; the disease was more severe when both subsets were transferred (Fig. 2c).
In the NOD mouse, males are more resistant to IDDM than females, due to an effect of male sex hormones [24,25]. Taking note of this gender dependency, we compared, on the one hand, the susceptibility of the target β-cells to autoimmune destruction and, on the other hand, the ability of the islet lymphocytes to cause the disease. We found that male islets were actually more susceptible to lymphocyte-mediated autoimmune damage than were female islets (Fig. 3c)—an observation that cannot explain why male NOD mice are less prone to the disease. However, male islet-infiltrating lymphocytes were less aggressive, even in the female recipient environment (Fig. 3a). One can conclude that the properties of the effector lymphocytes, rather than the susceptibility of the islets, determine the NOD sex difference. The molecular mechanism responsible for the greater sensitivity of the male β-cells to destruction remains to be investigated.
The lower pathogenicity of male islet lymphocytes was associated with a relatively lower content of IFN-γ-secreting T cells (Fig. 3b). Similarly, when female NOD mice were protected from IDDM by peptide p277 treatment, the ability of their islet lymphocytes to induce diabetes was inhibited (Fig. 4a), and there were fewer T cells capable of secreting IFN-γ than in the group treated with the control peptide, MTp278 (Fig. 4b).
In contrast, acceleration of IDDM by cyclophosphamide rendered the islet lymphocytes more aggressive: the disease progressed in the NOD.scid recipients more rapidly, and the numbers of IFN-γ SFC were higher than in the control group (Fig. 5a,b). Since NOD.scid mice treated with cyclophosphamide without cell transfer did not develop diabetes (data not shown), it is unlikely that acceleration of the disease was due to direct toxicity of the drug to β-cells. It has been suggested that cyclophosphamide-induced acceleration of diabetes may be due to a preferential depletion of suppressor T cells [12]. However, cyclophosphamide treatment of the recipients significantly inhibited the development of IDDM (Fig. 5c), indicating that cyclophosphamide-sensitive cells are present in the insulitis, and that these cells are pathogenic. One may also speculate that cyclophosphamide-sensitive islet lymphocytes can suppress β-cell killing, but may also assist in tissue-specific homing and therefore might be necessary for disease in our transfer system.
The major finding of this study is that lymphocytes of the insulitis lesion differ in their ability to induce diabetes upon passive transfer. Pathogenicity correlated with the relative frequencies of T cells capable of secreting IFN-γ upon activation. IFN-γ has been reported to be involved in β-cell destruction in vitro and in vivo [26–28]. Although IFN-γ knockout NOD mice can develop IDDM [29], this Th1 cytokine seems to be used for attacking the β-cells by the immune system of wild-type NOD mice.
Another important finding was inhibition of the adoptive IDDM transfer by splenic lymphocytes. The existence of regulatory T cells in the spleen of prediabetic NOD mice has been proposed earlier [30,31]. This suggests that the periphery may be critical in maintaining the insulitis in a non-destructive state. Indeed, for successful passive transfer of IDDM using spleen cells, the donor mice have to be diabetic and the recipient mice have to be immunodeficient (i.e. athymic, irradiated or scid), otherwise the regulatory lymphocytes will prevent the acquired disease. Similarly, elimination of peripheral T cells by cyclophosphamide results in rapid clinical diabetes. Spontaneously diabetic BB rats are lymphopenic [32], and their reconstitution with lymphocytes from diabetes-resistant rats provides protection [33]. In the BB model of IDDM, protection has been attributed to RT6.1+ T lymphocytes [34]. The nature of protective cells in the NOD mouse spleen is uncertain. We could not detect differences in IFN-γ/IL-4 ratios between splenic and islet T cells, but a shift towards Th2 responses in the spleen may involve other cytokines or may be restricted to a relatively small subpopulation of memory CD4+ T cells, as has been suggested recently [31]. In fact, the inhibition of diabetes may be mediated by antigen-specific splenic Th2 cells: in p277-treated NOD female mice a strong Th2 response to the peptide may be detected in the spleen, as judged by secretion of IL-4 and IL-10 [7]. Whether these Th2 cells invade the lesion or recruit pathogenic cells from the insulitis is not known, but appropriate stimulation of the periphery appears to be an attractive alternative to immunosuppression [4,35].
Acknowledgments
I.R.C. is the incumbent of the Mauerberger Chair of Immunology and the Director of the Robert Koch-Minerva Center for Research in Autoimmune Diseases. V.A. is the recipient of the Samara Jan Turkel Scholarship for Autoimmune Diseases. The work was supported by grants from the Minerva Foundation, the Tauro Foundation and Portman Pharmaceuticals, Inc.
References
- 1.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tocino Y. Breeding of a non-obese, diabetic strain of mice. Exp Anim. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 2.Bach J-F. Insulin-dependent diabetes mellitus as a β-cell targeted disease of immunoregulation. J Autoimmun. 1995;8:439–63. doi: 10.1016/0896-8411(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 3.Baxter AJ, Adams MA, Mandel TE. Comparison of high- and low-diabetes-incidence NOD mouse strains. Diabetes. 1989;38:1296–300. doi: 10.2337/diab.38.10.1296. [DOI] [PubMed] [Google Scholar]
- 4.Rabinovitch A. Immunoregulatory and cytokine imbalances in the pathogenesis if IDDM. Therapeutic intervention by immunostimulation. Diabetes. 1994;43:613–21. doi: 10.2337/diab.43.5.613. [DOI] [PubMed] [Google Scholar]
- 5.Prochazka M, Gaskins HR, Shultz LD, Leiter EH. The nonobese diabetic scid mouse: model for spontaneous thymomagenesis associated with immunodeficiency. Proc Natl Acad Sci USA. 1992;89:3290–4. doi: 10.1073/pnas.89.8.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elias D, Cohen IR. Treatment of autoimmune diabetes and insulitis in NOD mice with heat shock protein 60 peptide p277. Diabetes. 1995;44:1132–8. doi: 10.2337/diab.44.9.1132. [DOI] [PubMed] [Google Scholar]
- 7.Elias D, Meilin A, Ablamunits V, Birk OS, Carmi P, Konen-Waisman S, Cohen IR. Hsp60 peptide therapy of NOD mouse diabetes induces a Th2 cytokine burst and downregulates autoimmunity to various β-cell antigens. Diabetes. 1997;46:758–64. doi: 10.2337/diab.46.5.758. [DOI] [PubMed] [Google Scholar]
- 8.Elias D, Reshev T, Birk OS, van der Zee R, Walker MD, Cohen IR. Vaccination against autoimmune mouse diabetes with a T-cell epitope of the human 65-kDa heat shock protein. Proc Natl Acad Sci USA. 1991;88:3088–91. doi: 10.1073/pnas.88.8.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gotoh M, Maki T, Kiyoizumi T, Satomi S, Monako AP. An improved method for isolation of mouse pancreatic islets. Transplantation. 1985;40:437–8. doi: 10.1097/00007890-198510000-00018. [DOI] [PubMed] [Google Scholar]
- 10.Sedgwick JD, Holt PG. The ELISA-plaque assay for the detection and enumeration of antibody-secreting cells. J Immunol Methods. 1986;87:37–44. doi: 10.1016/0022-1759(86)90341-8. [DOI] [PubMed] [Google Scholar]
- 11.Harada M, Makino S. Promotion of spontaneous diabetes in non-obese diabetes-prone mice by cyclophosphamide. Diabetologia. 1984;27:604–6. doi: 10.1007/BF00276978. [DOI] [PubMed] [Google Scholar]
- 12.Yasunami R, Bach J-F. Anti-suppressor effect of cyclophosphamide on the development of spontaneous diabetes in NOD mice. Eur J Immunol. 1988;18:481–4. doi: 10.1002/eji.1830180325. [DOI] [PubMed] [Google Scholar]
- 13.Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14:619–33. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- 14.Charlton B, Mandel T. Progression from insulitis to β-cell destruction in NOD mouse requires L3T4+ T-lymphocytes. Diabetes. 1988;37:1108–12. doi: 10.2337/diab.37.8.1108. [DOI] [PubMed] [Google Scholar]
- 15.Shehadeh NN, LaRosa F, Lafferty KJ. Altered cytokine activity in adjuvant inhibition of autoimmune diabetes. J Autoimmun. 1993;6:291–300. doi: 10.1006/jaut.1993.1025. [DOI] [PubMed] [Google Scholar]
- 16.Hancock WW, Polanski M, Zhang J, Blogg N, Weiner HL. Suppression of insulitis in NOD mice by oral insulin administration is associated with selective expression of IL-4, IL-10, TGF-β and prostaglandin-E. Am J Pathol. 1995;147:1193–9. [PMC free article] [PubMed] [Google Scholar]
- 17.Bendelac A, Carnaud C, Boitard Bach JF. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates: requirement for both L3T4+ and Lyt-2+ T cells. J Exp Med. 1987;166:823–32. doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller BJ, Appel MC, O'Neil JJ, Wicker LS. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J Immunol. 1988;140:52–58. [PubMed] [Google Scholar]
- 19.Peterson JD, Haskins K. Transfer of diabetes in the NOD-scid mouse by CD4 T-cell clones. Diabetes. 1996;45:328–36. doi: 10.2337/diab.45.3.328. [DOI] [PubMed] [Google Scholar]
- 20.Pankewycz O, Strom TB, Rubin-Kelley VE. Islet-infiltrating T cell clones from non-obese diabetic mice that promote or prevent accelerated onset diabetes. Eur J Immunol. 1991;21:873–9. doi: 10.1002/eji.1830210403. [DOI] [PubMed] [Google Scholar]
- 21.Healey D, Ozegbe P, Arden S, Chandler P, Hutton J, Cooke A. In vivo activity and in vitro specificity of CD4+ Th1 and Th2 T cells derived from the spleens of diabetic NOD mice. J Clin Invest. 1994;95:2979–85. doi: 10.1172/JCI118006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 23.Serezze DV, Chapman HD, Varnum DS, Gerling I, Leiter EH, Shultz LD. Initiation of autoimmune diabetes in NOD/Lt mice is MHC class I-dependent. J Immunol. 1997;158:3978–86. [PubMed] [Google Scholar]
- 24.Fitzpatric F, Lepault F, Homo-Delarche F, Bach J-F, Dardenne M. Influence of castration, alone or in combination with thymectomy, on the development of diabetes in the nonobese diabetic mouse. Endocrinology. 1991;129:1382–90. doi: 10.1210/endo-129-3-1382. [DOI] [PubMed] [Google Scholar]
- 25.Fox HS. Androgen treatment prevents diabetes in nonobese diabetic mice. J Exp Med. 1992;175:1409–12. doi: 10.1084/jem.175.5.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pukel C, Baqueriso H, Rabinovitch A. Destruction of rat islet cell monolayers by cytokines. Synergistic interactions of interferon-γ, tumor necrosis factor, lymphotoxin, and interleukin 1. Diabetes. 1988;37:133–6. doi: 10.2337/diab.37.1.133. [DOI] [PubMed] [Google Scholar]
- 27.Debray-Sachs M, Carnaud C, Boitard C, Cohen H, Gresser I, Bedossa P, Bach J-F. Prevention of diabetes in NOD mice treated with antibody to murine IFN-γ. J Autoimmun. 1991;4:237–48. doi: 10.1016/0896-8411(91)90021-4. [DOI] [PubMed] [Google Scholar]
- 28.Rabinovitch A, Suares-Pinzon WL, Sorensen O, Bleackley RC, Power RF. IFN-γ gene expression in pancreatic islet-infiltrating mononuclear cells correlates with autoimmune diabetes in nonobese diabetic mice. J Immunol. 1995;154:4874–82. [PubMed] [Google Scholar]
- 29.Hultgren B, Huang X, Dybdal N, Stewart TA. Genetic absence of γ-interferon delays but does not prevent diabetes in NOD mice. Diabetes. 1996;45:812–7. doi: 10.2337/diab.45.6.812. [DOI] [PubMed] [Google Scholar]
- 30.Boitard C, Yasunami R, Dardenne M, Bach J-F. T cell-mediated inhibition of the transfer of autoimmune diabetes in NOD mice. J Exp Med. 1989;169:1669–80. doi: 10.1084/jem.169.5.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimada A, Rohane P, Fathman CG, Charlton B. Pathogenic and protective roles of CD45RBlow CD4+ cells correlate with cytokine profiles in the spontaneously autoimmune diabetic mouse. Diabetes. 1996;45:71–78. doi: 10.2337/diab.45.1.71. [DOI] [PubMed] [Google Scholar]
- 32.Jackson R, Rassi N, Crump T, Haynes B, Eisenbarth GS. The BB diabetic rat: profound T-cell lymphocytopenia. Diabetes. 1981;30:887–9. doi: 10.2337/diab.30.10.887. [DOI] [PubMed] [Google Scholar]
- 33.Rossini AA, Faustman D, Woda BA, Like AA, Szymanski I, Mordes JP. Lymphocyte transfusions prevent diabetes in the Bio-Breding/Worcester rat. J Clin Invest. 1984;74:39–46. doi: 10.1172/JCI111416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greiner DL, Mordes JP, Handler ES, Angelillo M, Nakamura N, Rossini AA. Depletion of RT6.1+ T lymphocytes induces diabetes in resistant biobreeding/Worcester (BB/W) rats. J Exp Med. 1987;166:461–75. doi: 10.1084/jem.166.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen IR. Treatment of autoimmune disease: to activate or to deactivate? Chem Immunol. 1995;60:150–60. [PubMed] [Google Scholar]