

Abstract
We have investigated the association between alleles of the genes for tumour necrosis factor-alpha (TNF-α) and TNF-β and severity of disease during malarial (Plasmodium falciparum) and other infections in the Sri Lankan population. Patients were categorized as having either (i) uncomplicated malaria, (ii) severe and complicated malaria, or (iii) severe and complicated infection in which a diagnosis of malaria had been excluded. For all the patients, as well as for a group of matched healthy controls, TNF-α and TNF-β allelic types were identified using the polymerase chain reaction (PCR) and allele-specific oligonucleotide probes and restriction enzyme digestion. The odds in favour of carrying the TNFα*2 allele, mainly of the heterozygous genotype (TNFα*1,*2), were two to three times greater among individuals with severe disease, of either malarial or other infectious origin, relative to healthy controls or to those with uncomplicated malarial infections. No significant risk was associated with either of the alleles of TNF-β.
Keywords: TNF polymorphisms, Plasmodium falciparum, malaria, infectious disease, pathogenesis
INTRODUCTION
Knowledge of the genetic basis of susceptibility to disease is an expanding field. Polymorphisms in genes within the MHC have been of primary interest in relation to diseases involving inflammatory and autoimmune disorders such as systemic lupus erythematosus (SLE) [1], rheumatoid arthritis (RA) [2] and insulin-dependent diabetes mellitus (IDDM) [3], among many others, and to infectious diseases such as malaria [4,5]. Previous studies of human malarial infection have identified associations between alleles or haplotypes of class I (B53) and class II (DRB1 *1302.DQB1*0501) MHC, or HLA, genes and reduced risk of severe and life-threatening disease during malarial infection in children in West Africa [4]. In the same population, homozygocity for the TNFα*2 allele was associated with greatly increased risk of severe (cerebral) disease and death during malarial infection [5]. Following these observations we have studied the distribution of polymorphisms at the TNF-α and also the TNF-β loci in relation to severe disease during malarial and other infections. We conducted these studies in a Sri Lankan population where malaria is prevalent at relatively low incidence compared with that in West Africa, but where it is, nevertheless, a common symptomatic infection among all age groups in a large part of the country.
TNF-α and TNF-β are cytokines which have close structural homology and about 30% amino acid sequence identity; both cytokines are recognized by the same widely distributed cellular TNF receptors and soluble TNF-binding proteins [6]. As a consequence, many of their numerous effects are similar, including a variety of pathologic effects in infectious and autoimmune diseases [7,8]. Their mechanisms of induction and their cellular sources are, on the other hand, different. TNF-α is the product of a wide variety of cells including macrophages, neutrophils, activated lymphocytes and natural killer (NK) cells among others, while TNF-β is produced only by lymphocytes. Induction and secretion of TNF-α is characteristically the result either of endotoxin-like interactions directly with monocytes and other circulating cells, or of activated lymphocytes involved in cognate recognition of antigen. In malaria, the resulting TNF-α can be involved in the generation of disease symptoms and pathology. TNF-α is, for example, a key endogenous pyrogen in fever responses to endotoxin-like substances, as has been verified in malarial infections [9,10]. There is also a substantial literature which associates the presence of TNF-α with various aspects of severe pathology in Plasmodium falciparum infection, including cerebral malaria and anaemia and renal, hepatic, pulmonary and circulatory failure [11]. TNF-β has been little studied in malarial infection and disease [12], but for the reasons given it can be expected that its effects could be related to those of TNF-α.
In addition to similarity in the function of these cytokines, there are reports which suggest that polymorphisms in the human genes for TNF-α and TNF-β mutually influence their production. Thus peripheral blood mononuclear cells (PBMC) stimulated with lipopolysaccharide (LPS) secreted more TNF-α when they were from individuals carrying the TNFβ*2 allele than from TNFβ*1*1 homozygotes [13]. In another study [14], PBMC from individuals carrying the TNFβ*2 allele secreted less TNF-β under stimulation with the mitogen phytohaemagglutinin (PHA) than from individuals homozygous for TNFβ*1*1. Yet again, PBMC from individuals carrying a TNFα*2 allele produced more of both TNF-α and TNF-β under the stimulation of T lymphocyte activators than did PBMC from individuals homozygous for TNFα*1,*1 [15]. Because of this evidence that different combinations of alleles at the TNF-α and TNF-β loci can affect cytokine secretion, we considered it important to include both loci in our investigation of the relationship between TNF polymorphisms and severity of disease in the present study.
The genes for TNF-α and TNF-β lie in tandem within a region of approx. 7 kb in the centre of the MHC locus on the short arm of human chromosome 6 between the HLA-B and the HLA-D loci [16,17]. In addition to numerous microsatellite polymorphisms described near the two TNF loci [18,19], several di-allelic point substitutions are known [14,20–23] (Fig. 1). These include a point substitution at position −308 (in relation to the TNF-α transcription start site) in the promoter region of TNF-α [22] which defines the TNF-α alleles *1 (G at −308) and *2 (A at −308). A di-allelic polymorphism in the TNF-β gene consists of three separate point substitutions [14]. Two of these occur in the first intron of the gene, one at nucleotide position +252 (from the transcriptional start site of the first exon of TNF-β gene) forming an NcoI restriction fragment length polymorphism (RFLP), the other being at position +368. The third point substitution is at position +723 in the third exon of the TNF-β gene and encodes a substitution at amino acid position 26 in the mature TNF-β protein. The substitutions at these three points define TNF-β alleles *1 and *2. Thus TNF-β allele *1 is defined by NcoI fragments of 5.7 and 5.3 kb and carries a G at nucleotide position +368 and asparagine at amino acid position 26 in the mature protein (A at nucleotide position +723) [14]. TNF-β allele *2 is defined by an NcoI fragment of 11 kb and carries a C at nucleotide position +368 and threonine at amino acid position 26 (C at nucleotide position +723) [14].
Fig. 1.
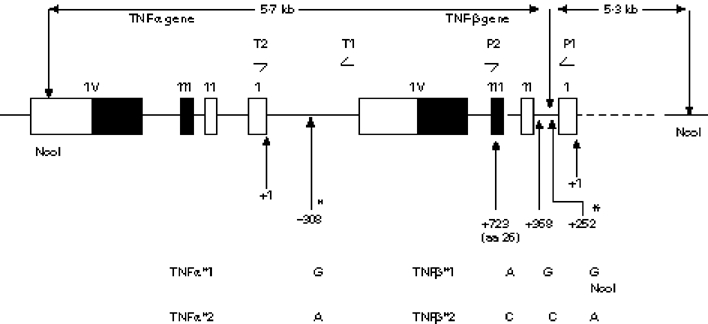
Schematic representation of the tandemly arranged TNF-α and TNF-β genes showing positions of diallelic polymorphisms. Open boxes and closed boxes represent untranslated and translated parts of the exons, respectively. Positions of the diallelic polymorphisms of the TNF genes are marked as −308 (TNF-α) and +723, +368, +252 (TNF-β) in relation to the transcription start sites of the two genes. The two polymorphisms which were used to type patients in this study are indicated as * and include the NcoI site polymorphism which distinguishes TNFβ*1 and *2. Positions of the oligonucleotide primers used to amplify the TNF-α and TNF-β genes are indicated as T1 and T2, and P1 and P2, respectively (see text).
We have analysed the association between different genotypes of TNF-α and TNF-β as defined by the polymorphisms described above, and severity of disease in P. falciparum malaria and other infections in the Sri Lankan population. As in the study by McGuire et al. [5] in a West African population, we found a strong association between increased risk of severe disease during malarial infection and presence of the TNFα*2 allele. In contrast to the previous study, however, this increased risk occurred in the heterozygous state (TNFα*1, *2) and not only in the homozygotes (TNFα*2,*2) as had been reported by McGuire et al. [5]. No significant risk of severe disease was found in association with either allele of TNF-β.
MATERIALS AND METHODS
Selection of human subjects
All patients were selected from the National Hospital of Sri Lanka, in the capital city, Colombo, and the General Hospital of Anuradhapura in the North-Central province of Sri Lanka. The inclusion criteria for this study for malaria patients of the uncomplicated (UC) and severe and complicated (SC) categories were a blood film positive for P. falciparum-infected erythrocytes and having no other coexisting illness. All SC patients had an altered level of consciousness, with, in 45% of them, other organ dysfunctions also being present. The categorization of patients into the UC and SC groups was done on the basis of an evaluation using a numerical scoring system (severe and complicated score) which was developed by us based on a combination of the original 14 accepted clinical criteria for severe and complicated malaria [24] and on the results of a series of biochemical and other laboratory investigations to assess the functional state of vital organs/systems [25].
The severe non-malarious category (SN) included patients who had impaired consciousness and were being managed in intensive care units of these hospitals as clinical states of infective origin, all cases with non- infective aetiologies, such as cerebrovascular accidents (CVA), diabetic coma, space occupying lesions and hepatic encephalopathies, having been excluded. All patients in the SN group were those in whom there had been a strong clinical suspicion of malaria until they were diagnosed as negative for malaria by repeated screening of thin and thick blood films, by the ParaSightTM-F dip-stick test (Becton Dickinson, Sparks, MD) and by a diagnostic polymerase chain reaction (PCR) [26]. All patients in this category gave a history of an illness of acute onset characterized by fever and headache preceding impairment of consciousness. Nine percent of SN patients gave a history of convulsions, 15% were subsequently diagnosed as having pyogenic meningitis on the basis of cerebrospinal fluid (CSF) examination, and a further 42% of cases were diagnosed as viral encephalitis following viral antibody tests. In the remaining 34% of cases a diagnosis could not be confirmed, although on clinical grounds they were diagnosed as having leptospirosis or other viral disease. The illness was fatal in 20% of this SN group, the rest making a complete recovery without apparent sequelae.
Collection of blood samples and extraction of DNA
Blood samples were collected from human subjects into microvette tubes (Sarsted, Germany) for extraction of host DNA. Blood samples were frozen at −20°C until they were used for DNA extractions. DNA was extracted from patients' blood using the standard phenol chloroform extraction method [27] and typed for TNF-α and TNF-β gene alleles as follows.
Allelic typing of the TNF-α gene
A fragment of 519 bp (from −502 to +17 from the transcription start site of the structural gene, see Fig. 1) from the promoter region of the human TNF-α gene, and which includes a single base polymorphism at −308, was amplified from the DNA samples by PCR with oligonucleotide primers T1 and T2 as used by McGuire et al. [5] (see Fig. 1). The TNF-α primer sequences were: T1–5′ CAA ACA CAG GCC TCA GGA CTC 3′; T2–5′ AGG GAG CGT CTG CTG GCT G 3′. The PCR amplified products were probed with allele-specific oligonucleotide probes by the Southern hybridization technique. The sequences of these allele-specific oligonucleotide probes, which were end-labelled with γ32P, were, for TNFα*1–5′ AGG GGC ATG GGG ACG GG 3′, and for TNFα*2–5′ AGG GGC ATG AGG ACG GG 3′.
Allelic typing of the TNF-β gene
A 740bp fragment from the ′5 non-coding sequence (from +52 to +792 from the transcription start site of the TNF-β gene, see Fig. 1) was amplified by PCR from human DNA samples. The primer sequences used were −5′ CCC GTG CTT CGT GCT TTG GAC TA 3′ (P1, see Fig. 1) and −5′ AGA GCT GGT GGG GAC ATG TCT G 3′ (P2, see Fig. 1) as used by Campbell et al. [28]. The amplified products of the TNF-β gene were then analysed for RFLP by digesting with the NcoI restriction enzyme and separation on agarose gel electrophoresis. Fragments of 540 bp and 200 bp indicated the presence of the TNFβ*1 allele, and the full length fragment (740 bp) indicated the presence of the TNFβ*2 allele.
Statistical analysis
Experimental data for the distribution of TNF-α and TNF-β alleles and clinical data were entered in a computer database and comparisons were made by the χ2 tests, t-tests and normal theory tests using either the EPIINFO, SPSS or EPISTAT statistical software packages.
RESULTS
We studied groups of malaria patients in Sri Lanka suffering either from uncomplicated infections or from severe and complicated infections of P. falciparum as defined by the criteria referred to in Materials and Methods, together with a group of patients suffering from severe disease defined according to the same clinical and biochemical criteria, but having malaria excluded as a diagnosis. Three disease groups— uncomplicated malaria (UC), severe and complicated malaria (SC), severe non-malaria (SN) and a group of healthy control individuals (HC)—were characterized for TNF-α and TNF-β genotypes as described in Materials and Methods.
The frequencies of alleles of the genes for TNF-α and TNF-β in disease groups and in healthy controls (Table 1)
Table 1.
Frequencies of alleles of TNF-α and TNF-β among individuals in disease groups and in healthy controls
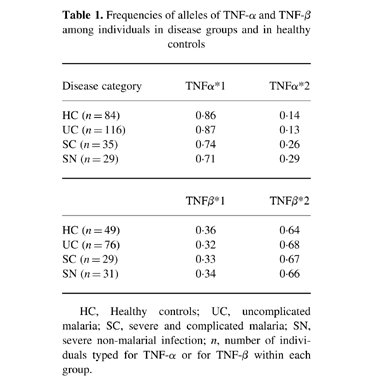
HC, Healthy controls; UC, uncomplicated malaria; SC, severe and complicated malaria; SN, severe non-malarial infection; n, number of individuals typed for TNF-α or for TNF-β within each group.
The frequencies in the Sri Lankan population at large (healthy controls) of alleles *1 and *2 of the gene for TNF-α (TNFα*1 and TNFα*2) were 0.86 and 0.14, respectively, which is similar to their frequencies as reported in a European (Dutch) population [29]. The frequency of TNFα*2, at 0.14, was slightly higher than that found in a West African population, 0.11 [5].
Within the Sri Lankan population no difference was found in the frequencies of the TNFα*1 and TNFα*2 alleles between the HC and UC groups (Table 1). However, within the severe groups, both malarial (SC) and non-malarial (SN), the frequencies of TNFα*2, at 0.26 and 0.29, respectively, were significantly higher than in the healthy control group (P = 0.02 and 0.05, respectively).
The frequencies of the two TNF-β alleles were very similar in all four Sri Lankan groups (HC, UC, SC and SN) at about 0.33 and 0.67 for TNFβ*1 and TNFβ*2, respectively (Table 1) and very similar to the reported frequencies of these alleles in a European population [29].
The association of genotypes of the genes for TNF-α and TNF-β with severe disease (Table 2)
Table 2.
Numbers of individuals in the different categories for disease (see Table 1) divided according to genotype for TNF-α and TNF-β
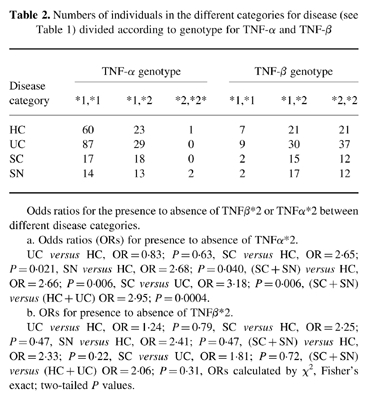
Odds ratios for the presence to absence of TNFβ*2 or TNFα*2 between different disease categories.
a. Odds ratios (ORs) for presence to absence of TNFα*2.
UC versus HC, OR = 0.83; P = 0.63, SC versus HC, OR = 2.65; P = 0.021, SN versus HC, OR = 2.68; P = 0.040, (SC + SN) versus HC, OR = 2.66; P = 0.006, SC versus UC, OR = 3.18; P = 0.006, (SC + SN) versus (HC + UC) OR = 2.95; P = 0.0004.
b. ORs for presence to absence of TNFβ*2.
UC versus HC, OR = 1.24; P = 0.79, SC versus HC, OR = 2.25; P = 0.47, SN versus HC, OR = 2.41; P = 0.47, (SC + SN) versus HC, OR = 2.33; P = 0.22, SC versus UC, OR = 1.81; P = 0.72, (SC + SN) versus (HC + UC) OR = 2.06; P = 0.31, ORs calculated by χ2, Fisher's exact; two-tailed P values.
The above data indicated an association between the presence of the TNFα*2 allele and severe disease during malarial and other infections. In the study by McGuire et al. in a West African population [5], the TNFα*2,*2 homozygous state was associated with a high (approx. seven-fold increase) relative risk of severe (cerebral) malaria, but no such increased risk was associated with the TNFα*1,*2 heterozygous state. In contrast to these findings from West Africa, in the Sri Lankan population we found that the heterozygous combination, TNFα*1,*2, was two- to three-fold more common in the severe disease groups, both malarial (odds ratio (OR) = 2.76; P = 0.013) and non-malarial (OR = 2.42; P = 0.044) infections relative to the healthy controls. Although the number of TNFα*2,*2 homozygotes in our study was too low for statistical analysis, the two TNFα*2,*2 homozygotes among the infected individuals both had severe disease (non-malarial), which is consistent with a strong association of this genotype with severe disease (Table 2).
Carriers of the TNFα*2 allele, in either homozygous or heterozygous combination (TNFα*2 present), were significantly associated with severe disease, either malarial (SC) or non-malarial (SN), at ORs of between 2 and 3 relative to healthy controls (Table 2a). There was, however, no association of TNFα*2 with uncomplicated malarial infection.
The TNFβ*2 allele, in either homozygous or heterozygous combination (TNFβ*2 present), was also consistently associated with severe disease cases (malarial and non-malarial) at ORs similar to those for TNFα*2 (Table 2b), but the associations were not statistically significant.
DISCUSSION
We have found that the frequency in the Sri Lankan population studied here of allele *2 of the TNF-α gene (TNFα*2) was more than twice as high among individuals experiencing severe infectious disease, including malaria, as in the healthy population at large or among individuals experiencing clinically mild malarial infections. There was also an increased frequency of the TNFβ*2 allele among the severe disease groups relative to the mild disease groups and healthy controls, although the differences were not statistically significant.
Although our sample size was too small to draw statistically significant conclusions on the effects of TNF-β alleles on predisposition to severe infectious disease, we noted some interesting trends in our own data and those of others that we believe should be investigated further. In our study individuals who carried both a TNFα*2 and a TNFβ*2 allele were over-represented in the severe disease groups relative to individuals who lacked either one of these alleles (data not shown). At the same time this combination was under-represented, relative to the expectation from random reassortment of alleles, in the mild and control groups. Although our methods did not allow us to determine haplotype combinations of the alleles of TNF-α and TNF-β, this was done in the study by Bouma et al. [29] on a Dutch population. They found that all possible combinations of alleles of TNFα*1 and *2 and TNFβ*1 and *2 were represented in their study population with the exception of the TNFα*2/TNFβ*2 combination, which was totally absent. We suggest that these observations taken together could suggest that the TNFα*2/TNFβ*2 combination may be severely detrimental. If this is true it could account for the exactly contiguous arrangement on the chromosome of the genes for these two cytokines. This is because in such an arrangement, recombination events will be extremely rare and a detrimental haplotype, as we speculate TNFα*2/TNFβ*2 might be, could be virtually excluded from a population by its rapid elimination on the very rare occasions upon which it would arise. There would be no such defence, however, against the inheritance of a detrimental combination of alleles whose loci were far from each other on the same or separate chromosomes, because recombination between the loci could constantly reform the detrimental combination.
If this hypothesis is correct, then for both alleles at these two loci to be maintained within a human population there would have to be compensating advantages associated with each allele. Such advantage, at least for TNFα*2, is suggested by the study by Bouma et al. [29], in which this allele was significantly under-represented, by about one third, among patients with ulcerative colitis.
An association between high risk of severe malarial disease and presence of the TNFα*2 allele was previously reported by McGuire et al. [5] in a study conducted on children in West Africa. In their study, however, the higher risk of severe disease was found only among TNFα*2,*2 homozygotes and not, as in the present study, among heterozygotes for the TNFα*2 allele (TNFα*1,*2). There could be several explanations for this difference. There are, for example, large differences in the epidemiology and the pathology of malarial infections in West Africa and in Sri Lanka. In West Africa high levels of immunity to malaria are achieved relatively early in life due to the intense rates of malaria transmission which prevail there, and malarial disease, including almost all severe and fatal infections of malaria, is found predominantly in young children. In Sri Lanka, on the other hand, malaria inoculation rates are too low to induce more than moderate to very low levels of age/exposure-acquired immunity, and malarial infections, including severe disease, occur with similar frequency at all ages. This difference in levels of endemicity of malaria is probably the primary reason for the differences in the types and distributions of severe malarial disease seen in the two regions. The severe disease associated with TNFα*2,*2 homozygosity in the West African study [5] was almost exclusively cerebral malaria, and because of early, exposure-acquired, immunity to malaria was only in young children. In the Sri Lankan populations, on the other hand, a high proportion of severe malarial infections is in adults and manifests more commonly as MODS (Multiple Organ Dysfunction Syndrome), the rest being cerebral malaria. Such differences in levels of immunity and the age at which severe malarial disease occurs in a population could, perhaps, lead to the expression of different disease manifestations for the same genetic make-up of the host.
Another possibility is that differences in genetic background between the West African and Sri Lankan populations determine the different effects of a specific allele, such as TNFα*2, on the outcome of malarial disease. If, as speculated here, the TNFα*2,β*2 combination is indeed detrimental during infectious disease, then a low frequency of the TNFβ*2 allele in the West African populations could be sufficient to account for observed differences in the associations between TNF-α genotype and disease severity in these two populations. In this context information on TNF-β allele frequencies and their involvement as risk factors in disease severity in West African populations would clearly be of great interest.
Acknowledgments
This work was supported by a grant from the WHO/UNDP/World Bank Special Programme for Research and Training in Tropical Diseases.
References
- 1.Wilson AG, Gordon C, di Giovine FS, de Vries N, van der Putte LBA, Emery P, Duff GW. A genetic association between systemic lupus erythematosus and tumour necrosis factor alpha. Eur J Immunol. 1994;24:191–5. doi: 10.1002/eji.1830240130. [DOI] [PubMed] [Google Scholar]
- 2.Stastny P. Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. New Eng J Med. 1978;298:869–71. doi: 10.1056/NEJM197804202981602. [DOI] [PubMed] [Google Scholar]
- 3.Svejgaard A, Jakobsen BK, Platz P, et al. HLA associations in insulin-dependent diabetes: search for heterozygosity in different groups of patients from a homogeneous population. Tissue Antigens. 1986;28:237–44. doi: 10.1111/j.1399-0039.1986.tb00489.x. [DOI] [PubMed] [Google Scholar]
- 4.Hill AVS, Allsopp CEM, Kwiatkowski D, et al. Common West African HLA antigens are associated with protection from severe malaria. Nature. 1991;352:595–600. doi: 10.1038/352595a0. [DOI] [PubMed] [Google Scholar]
- 5.McGuire W, Hill AVS, Allsopp CEM, Greenwood BM, Kwiatkowski D. Variation in the TNF-α promoter region associated with susceptibility to cerebral malaria. Nature. 1994;371:508–11. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 6.Smith CA. The TNF receptor superfamily of cellular and viral proteins—activation, costimulation and death. Cell. 1994;76:959–62. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 7.Grau GE, Lambert PH, Vassalli P, Piguet PF. Tumour necrosis factor cachectin as an effector of T-cell dependent immunopathology. Int Rev Exp Pathol. 1993;34(Part B):159–71. doi: 10.1016/b978-0-12-364935-5.50016-x. [DOI] [PubMed] [Google Scholar]
- 8.Beutler B, Cerami A. The biology of cachectin/TNF—a primary mediator of the host response. Ann Rev Immunol. 1989;7:625–55. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 9.Kwiatkowski D, Molyneux ME, Stephens S. Anti-TNF therapy inhibits fever in cerebral malaria. Quart J Med. 1993;86:91–98. [PubMed] [Google Scholar]
- 10.Karunaweera ND, Grau GE, Gamage CP, Carter R, Mendis KN. Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivax malaria. Proc Natl Acad Sci USA. 1992;89:3200–3. doi: 10.1073/pnas.89.8.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendis KN, Carter R. Clinical disease and pathogenesis in malaria. Parasitol Today. 1995;11(Suppl.):1–16. doi: 10.1016/0169-4758(95)80143-x. [DOI] [PubMed] [Google Scholar]
- 12.Clark IA, Gray KM, Rockett EJ, Cowden WB, Rockett KA, Ferrante A, Aggarwal BB. Increased lymphotoxin in human malarial serum and the ability of this cytokine to increase plasma interleukin-6 and cause hypoglycaemia in mice: implications for malarial pathology. Trans Roy Soc Trop Med Hyg. 1991;86:602–7. doi: 10.1016/0035-9203(92)90144-2. [DOI] [PubMed] [Google Scholar]
- 13.Pociot F, Briant L, Jongeneel CV, et al. Association of tumor necrosis factor (TNF) and class II major histocompatibility complex alleles with the secretion of TNF-α and TNF-β by human mononuclear cells: a possible link to insulin-dependent diabetes mellitus. Eur J Immunol. 1993;23:224–31. doi: 10.1002/eji.1830230135. [DOI] [PubMed] [Google Scholar]
- 14.Messer G, Spengler U, Jung MC, Honold G, Blomer K, Pape GR, Riethmuller G, Weiss EH. Polymorphic structure of the tumor necrosis factor (TNF) locus: an Ncol polymorphism in the first intron of the human TNF-β gene correlates with a variant amino acid in position 26 and a reduced level of TNF-β production. J Exp Med. 1991;173:209–19. doi: 10.1084/jem.173.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouma GB, Crusius JBA, Oudkerk Pool M, et al. Secretion of tumour necrosis factor α and lymphotoxin α in relation to polymorphisms in the TNF genes and HLA-DR alleles. Relevance for inflammatory bowel disease. Scand J Immunol. 1996;43:456–63. doi: 10.1046/j.1365-3083.1996.d01-65.x. [DOI] [PubMed] [Google Scholar]
- 16.Nedospasov SA, Shakov AN, Turetskaya RL, et al. Tandem arrangement of genes coding for tumor necrosis factor (TNF-α) and lymphotoxin (TNF-β) in the human genome. Cold Spring Harbor Symp Quant Biol. 1986;51:611–24. doi: 10.1101/sqb.1986.051.01.073. [DOI] [PubMed] [Google Scholar]
- 17.Spies T, Morton CC, Nedospasov SA, Fiers W, Pious D, Strominger JL. Genes for tumor necrosis factor α and β are linked to the human major histocompatibility complex. Proc Natl Acad Sci, USA. 1986;83:8699–702. doi: 10.1073/pnas.83.22.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jongeneel CV, Briant L, Udalova IA, Sevin A, Nedospasov SA, Cambon-Thomsen A. Extensive genetic polymorphism in the human tumor necrosis factor region and relation to extended HLA haplotypes. Proc Natl Acad Sci, USA. 1991;88:9717–21. doi: 10.1073/pnas.88.21.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nedospasov SA, Udalova IA, Kuprash DV, Turetskaya RL. DNA sequence polymorphism at the human tumor necrosis factor (TNF) locus: numerous TNF/lymphotoxin alleles tagged by two closely linked microsatellites in the upstream region of the lymphotoxin (TNF-β) gene. J Immunol. 1991;147:1053–9. [PubMed] [Google Scholar]
- 20.Abraham LJ, Du DC, Zahedi K, Dawkins RL, Whitehead AS. Haplotypic polymorphisms of the TNFB gene. Immunogenetics. 1991;33:50. doi: 10.1007/BF00211695. [DOI] [PubMed] [Google Scholar]
- 21.Ferencik S, Lindemann M, Horsthemke B, Grosse-Wilde H. A new restriction fragment length polymorphism of the human TNF-B gene detected by Asp HI digest. Eur J Immunogenetics. 1992;19:425–30. doi: 10.1111/j.1744-313x.1992.tb00086.x. [DOI] [PubMed] [Google Scholar]
- 22.Wilson AG, di Giovine FS, Blakmore AIF, Duff GW. Single base change in the human tumour necrosis factor alpha (TNFα) gene detected by Nco1 restriction of PCR product. Hum Mol Genecs. 1992;1:353. doi: 10.1093/hmg/1.5.353. [DOI] [PubMed] [Google Scholar]
- 23.D'Alfonso S, Richiardi PM. A polymorphic variation in a putative regulation box of the TNFA promoter region. Immunogenetics. 1994;39:150–4. doi: 10.1007/BF00188619. [DOI] [PubMed] [Google Scholar]
- 24.Warrell DA, Molyneux ME, Beals PF. Severe and Complicated malaria. Trans Roy Soc Trop Med Hyg. 1990;84(Suppl. 2):1–65. [PubMed] [Google Scholar]
- 25.Alles HK. University of Colombo; 1998. PhD Thesis. [Google Scholar]
- 26.Snounou G, Viriyakosol S, Zhu XP, Jarry W, Pinheiro L, Rosario VE, Thaithong S, Brown KN. High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol. 1993;61:315–20. doi: 10.1016/0166-6851(93)90077-b. [DOI] [PubMed] [Google Scholar]
- 27.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 28.Campbell DA, Nelson S, Madhok R, Field M, Gallagher G. TNF Nco-I RFLP is not an independent risk factor in rheumatoid arthritis. Eur J Immunol. 1994;21:461–7. doi: 10.1111/j.1744-313x.1994.tb00219.x. [DOI] [PubMed] [Google Scholar]
- 29.Bouma G, Xia B, Crusius JBA, Bioque G, Koutroubakis I, Von Blomberg BME, Meuwissen SGM, Pena AS. Distribution of four polymorphisms in the tumour necrosis factor (TNF) genes in patients with inflammatory bowel disease (IBD) Clin Exp Immunol. 1996;103:391–6. doi: 10.1111/j.1365-2249.1996.tb08292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]