

Abstract
Immunosuppressive therapy with methotrexate (MTX) has been established as effective treatment for patients with rheumatoid arthritis. To analyse the therapeutic potential and mechanisms of action of MTX, we determined serum cytokine levels and cytokine production by splenic T cells and macrophages in untreated and MTX-treated mice. Furthermore, we assessed the role of MTX in a murine model of experimental arthritis induced by collagen type II (CIA). MTX reduced spontaneous and IL-15-induced tumour necrosis factor (TNF) production by splenic T cells but not by macrophages from healthy mice in vitro in a dose-dependent manner. In contrast, interferon-gamma (IFN-γ) production was less strikingly reduced and IL-4 production was virtually unaffected. In addition, treatment of healthy mice with MTX in vivo led to reduced TNF serum levels and diminished TNF production by splenic T cells and macrophages. Intraperitoneal administration of MTX prior to the onset of arthritis completely prevented clinical and pathological signs of CIA. This was associated with a striking reduction of TNF production by spleen cells from MTX-treated mice. The role of TNF in MTX-mediated effects on cytokine production was further underlined by the finding that MTX effects on IFN-γ production were augmented in TNF-transgenic mice but abrogated in mice in which the TNF-α gene had been inactivated by homologous recombination. Thus, MTX specifically modulates spontaneous and IL-15-induced TNF-α production in mice and prevents experimental murine CIA. These data suggest that TNF production by T cells is an important target of MTX and may serve as a basis to understand and further analyse MTX-mediated mechanisms of immunosuppression in patients with RA.
Keywords: methotrexate, cytokines, arthritis, tumour necrosis factor
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic, systemic disorder that is characterized by a symmetric polyarthritis finally leading to progressive joint destruction [1]. Keystones of RA are joint inflammation, proliferation of synovial cells, and attachment and invasion of synovial fibroblasts into adjacent cartilage and bone, mediated by continuous release of matrix-degrading enzymes [2]. Although the aetiology of RA remains undefined, a large body of data suggests that T lymphocytes, macrophages, and proliferating synovial cells are very likely to play a major role in the pathogenesis of this disease [2–4]. In particular, cytokines produced by these cells appear to be critically involved in the initiation and perpetuation of RA [3]. For instance, macrophage- and fibroblast-derived cytokines such as IL-1, IL-6 and tumour necrosis factor-alpha (TNF-α) are reproducibly found in both RA synovial fluid and synovial membrane [1–3]. These proinflammatory cytokines are considered to play a prominent role in RA pathogenesis by modulating cell migration, T and B cell activation, cartilage destruction and synovial proliferation [2,5]. Recent studies by Elliott et al. have shown that regulation of cytokine levels in patients with RA may also be a novel approach for treatment of these diseases [6,7]. They described successful treatment of refractory arthritis in patients with RA by i.v. infusion of antibodies to TNF-α, suggesting a key role for this cytokine in the pathogenesis of chronic arthritis.
TNF-α is a member of the TNF family of proteins that plays an important role in antibacterial host defence, contact hypersensitivity reactions, endotoxic shock, experimental colitis and arthritis, induction of fever and stimulation of production of several acute-phase proteins [8–13]. In RA, TNF is produced by most of the cells present in rheumatoid synovium, including fibroblasts, macrophages, endothelial cells and lymphocytes, and has been suggested to play a major pathogenic role [14].
Among the disease-modifying anti-rheumatic drugs (DMARDs), methotrexate (MTX) has become the predominant immunosuppressive treatment for patients with RA [5,15,16]. However, its mechanism of action has been only partially characterized. MTX is a folic acid antagonist that competitively inhibits dihydrofolate reductase (reviewed in [5,15]). It acts mainly on actively proliferating cells in the S phase of proliferation. In addition to its inhibitory effect on B cell function, MTX also suppresses macrophage function, modulates IL-1 and superoxide anion production and inhibits neutrophil chemotaxis [5,17–21]. Furthermore, it inhibits the binding of IL-1β to its receptor on peripheral blood cells [22] and it has anti-inflammatory activities by induction of cellular adenosine release [5]. In the present study, we asked whether MTX has specific effects on proinflammatory cytokine production in vitro and in healthy and arthritic mice in vivo. We demonstrate that MTX specifically modulates spontaneous and induced TNF production in mice and prevents experimental murine CIA. These data may serve as a basis to understand and further analyse MTX-mediated mechanisms of immunosuppression in patients with RA.
MATERIALS AND METHODS
Mice
BALB/c and DBA/1 mice were obtained from Harlan Laboratories (Borchen, Germany). The generation and characterization of TNF-α knockout mice has been described [23]. Screening for homozygous mice was performed as described [23]. Tg197 transgenic mice expressing transmembrane TNF protein have been described [12]. Mice were given PBS or indicated amounts of MTX (obtained from Sigma, Munich, Germany) by intraperitoneal injection (total injection volume 200 μl).
Induction of CIA
Male DBA/1 mice were obtained from Harlan Labs. Water and food were provided ad libitum. For induction of severe CIA, mice were primed with 500 μl pristane (Roth, Munich, Germany) as previously described [24,25] and immunized (at the age of 10–12 weeks) at day 21 by administration of 100 μg collagen type II (CII; Sigma) in Freund's complete adjuvant (FCA). Booster injections with 50 μg CII in FCA were given 3 and 6 weeks later.
Materials
FCA was obtained from Difco (Detroit, MI). Purified hamster anti-mouse CD3ε (clone 145-2C11) and hamster anti-mouse CD28 (clone 37.51) antibodies were obtained from Pharmingen (Hamburg, Germany). LPS (bacterial lipopolysaccharide), folic acid, phytohaemagglutinin (PHA) and phorbol 12-myristate 13-acetate (PMA) were obtained from Sigma. Recombinant simian IL-15 free of endotoxins was obtained from Genzyme (Rüsselheim, Germany) and was used at indicated concentrations.
Assessment of arthritis
Mice were examined visually for the appearance of arthritis in the peripheral joints and disease severity was graded on a scale, as specified below. Mice were considered to have arthritis when significant changes in redness and/or swelling were noted in the digits or in other parts of the paws. Arthritis was graded semiquantitatively on a scale of 0–4 for each paw: 0 = no changes; 0.5 = significant swelling and redness of one digit; 1 = swelling and erythema of two digits; 2 = mild swelling and erythema of the limb or swelling of more than two digits; 3 = marked swelling and erythema of the limb; and 4 = maximal swelling and redness of the limb and later, ankylosis. The macroscopic score (mean ± s.d.) was expressed as a cumulative value for all paws, with a maximum possible score of 16.
Grading of histological changes
Tissues were removed from the DBA mice with CIA at indicated time points and fixed in 10% formic acid for at least 12 h. Decalcification of samples was then performed in EDTA. Paraffin sections (4 μm) were made and stained with haematoxylin and eosin. Joint lesions on microscopic sections were graded semiquantitatively from 0 to 3, as previously described [26]. The following criteria were analysed: (i) synovial lesions: 0, no lesions, 1, mild effect, 2, moderate effect/proliferation, 3, severe lesions with destruction; (ii) inflammatory infiltrate: 0, none, 1, mild infiltrate, 2, moderate infiltrate, 3, severe infiltrate; (iii) cartilage destruction: 0, none, 1, mild, 2, moderate, 3, severe destruction with loss or complete fragmentation of cartilage; (iv) bone destruction: 0, none, 1, mild destruction of subchondral bone, 2, moderate destruction, 3, severe destruction with loss of large areas of bone; (v) bone neoformation: 0, none, 1, mild, 2, moderate, 3, extensive bone neoformation. Grading was done in a blinded fashion by the same pathologist (P.S.).
Cell isolation and purification of spleen mononuclear cells
Spleen mononuclear cells were isolated from freshly obtained specimens. In brief, the resected spleen was washed thoroughly in Hanks' balanced salt solution (HBSS) free of calcium and magnesium. The tissue was cut into small pieces and pushed through a 40-μm nylon cell strainer (Falcon, Becton Dickinson Labware, Heidelberg, Germany). Erythrocytes were removed from the resulting cell suspension by hypotonic lysis in ACK buffer. Spleen cells were then washed in PBS, centrifuged at 800 g for 10 min at 4°C and resuspended as specified below.
Purification of CD4+ T lymphocytes using immunomagnetic beads
In some experiments, immunomagnetic beads specific for CD4+ (obtained from Miltenyi, Hamburg, Germany) were used to isolate CD4+ T cells by MACS sorting [27]. Only cell populations with a purity >95% (as assessed by FACS analysis) were used in the experiments described below. The cells were cultured in a humidified atmosphere with 5% CO2 in a 37°C incubator.
Isolation of macrophage-enriched spleen cells
Macrophage-enriched cells were isolated from spleen mononuclear cells. The latter cells were depleted of CD4+ T cells by immunomagnetic beads as described above and plated on 35-cm plastic tissue culture dishes (Falcon Labware, Oxnard, CA) in 12 ml RPMI 1640 for 60 min at 37°C. Non-adherent cells were washed free with PBS and adherent cells were put on ice and washed free with two rinses of prewarmed PBS. Macrophage-enriched cells were >75% Mac1+ as assessed by FACS analysis.
Cell culture of spleen cells
Cell cultures of splenic mononuclear cells were performed in complete medium consisting of RPMI 1640 (Whittaker, Walkersville, MD) supplemented with 3 mml-glutamine, 10 mm HEPES buffer, 100 U/ml each of penicillin and streptomicin (Whittaker), and 10% heat-inactivated fetal calf serum (FCS). Cells were resuspended in complete RPMI media and cultured in complete media for 48 h.
Isolation of mRNA and reverse transcriptase- polymerase chain reaction
Total RNA of spleen cells was prepared using Qiagen RNeasy columns (Qiagen, Hilden, Germany) and reverse transcription of RNA into complementary DNA was made using MMLV reverse transcriptase (Boehringer, Mannheim, Germany). Polymerase chain reaction (PCR) was performed with murine TNF- and β-actin-specific primers derived from previously published sequence data [11]: TNF, 5′-GAG TGA CAA GCC TGT AGC CC-3′ and 5′-ATG GAT GAA CAC CCA TTC CC-3′; β-actin, 5′-TGA CGG GGT CAC CCA CAC TGT GCC CAT CTA-3′ and 5′-CTA GAA GCA TTT GCG GTG GAC GAT GGA GGG-3′. Cycling conditions were as follows: 94°C for 30 s, 57°C for 30 s and 72°C for 75 s for 25 cycles, followed by 10 min at 72°C for final extension. PCR products were analysed on 2% agarose gels.
Cytokine assays
To measure cytokine production, 24-well plates (Cellstar; Greiner, Frickenhausen, Germany) were coated with 10 μg/ml murine anti-CD3ε antibody in carbonate buffer pH 9.6 overnight at 4°C. Spleen mononuclear cells (10 × 106) were then cultured in 1 ml of complete medium in precoated or uncoated wells and 1 μg/ml soluble anti-CD28 antibody was added to the anti-CD3-coated wells. In addition, 40 μg/ml LPS and 50 ng/ml PMA were added to the wells, as specified in Results. Cultures were incubated at 37°C in a humidified incubator containing 5% CO2. After 48 h, culture supernatants from unstimulated and stimulated cells were removed and assayed for cytokine concentration.
Cytokine concentrations were determined by specific ELISA. Monoclonal rat anti-mouse cytokine antibodies were diluted at 2000 ng/ml in carbonate buffer pH 9.6. Antibody solution (50 μl per microwell) was then aliquoted into 96-well microtitre plates (Costar, Cambridge, MA). Plates were incubated at 4°C overnight, washed three times with PBS–Tween solution, and blocked for 1 h with 3% bovine serum albumin (BSA) in PBS. At this point, 50 μl of sample or standards of recombinant murine cytokines (Genzyme, Pharmingen, R&D Systems, Wiesbaden, Germany) were added to the wells and allowed to incubate at room temperature for 2 h. Plates were then washed four times, after which 50 μl of biotin-labelled rat anti-mouse cytokine antibody (2 μg/ml) in PBS with 0.1% BSA were added to each well and incubated at room temperature for 60 min. Plates were then washed an additional four times, after which 75 μl of a 1:1000 diluted solution of horseradish peroxidase (HRP)–streptavidin (Dako, Glostrup, Denmark) in PBS with 0.1% BSA were added to each well. Plates were then incubated for 30 min at room temperature and washed four times, after which 200 μl of 3,3′,5,5′-tetramethylbenzidine (Fluka, Buchs, Switzerland) were added to each well. Optical densities (OD) were measured on an EMAX ELISA reader (Molecular Division, Menlo Park, CA) at a wavelength of 450 nm.
Proliferation assays
Proliferation of cells was assessed by measuring 3H-TdR incorporation. In brief, 1 × 106 cells/ml were cultured in the presence of 50 ng/ml PMA and 10 μg/ml PHA in 96-well plates (Greiner) for 36 h; during the last 12 h of culture, 1 μCi of 3H-TdR (New England Nuclear, Boston, MA; specific activity 6.7 Ci/mmol) was added to each well; incorporated 3H radioactivity was measured in a scintillation counter (LS2800; Beckman Instruments, Inc., Fullerton, CA). Each proliferation assay was done in triplicate.
Statistical analysis
Significance of differences was assessed by Student's t-test using the program StatWorks.
RESULTS
MTX suppresses TNF and IFN-γ production by splenic mononuclear cells and CD4+ T lymphocytes in vitro
MTX effects on cytokine production in vitro and in vivo have only been partially characterized. In an initial series of studies, we therefore wanted to determine whether MTX can specifically modulate cytokine production by murine cells. Accordingly, we isolated splenic mononuclear cells from healthy BALB/c and DBA/1 mice and assayed their ability to produce proinflammatory cytokines in the presence and absence of MTX. In this experimental system, stimulation of murine spleen cells with anti-CD3/CD28, PMA and LPS led to strong production of proinflammatory cytokines such as TNF whose production peaked at 48 h (Fig. 1a). As shown in Fig. 1b, 0.1–10 μg/ml MTX significantly (P < 0.01) suppressed TNF-α production by splenic mononuclear cells from both BALB/c and DBA/1 mice after 48 h in a dose-dependent manner. However, production of TNF-α was more strikingly reduced compared with that of IL-1β, although levels of the latter cytokine were also significantly (0.1–10 μg/ml; P < 0.05) reduced upon MTX treatment in BALB/c mice. Changes of TNF production by spleen cells were already noted at low dosages (0.01–0.001 μg/ml) of MTX that did not suppress cell proliferation (Fig. 1b). This effect could be partially inhibited by increasing dosages of folic acid (Fig. 1c). Furthermore, there was no decrease of TNF mRNA levels after MTX treatment (Fig. 1d), suggesting that suppression of TNF production by MTX in primary spleen cells is not mainly due to inhibition of TNF gene transcription. Finally, production of IL-6 and IL-4 was not reduced by MTX treatment (Fig. 1b), indicating that MTX can selectively modulate cytokine production by mononuclear cells in vitro.
Fig. 1.
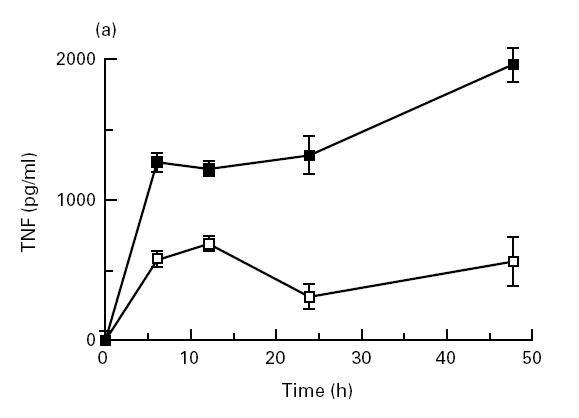
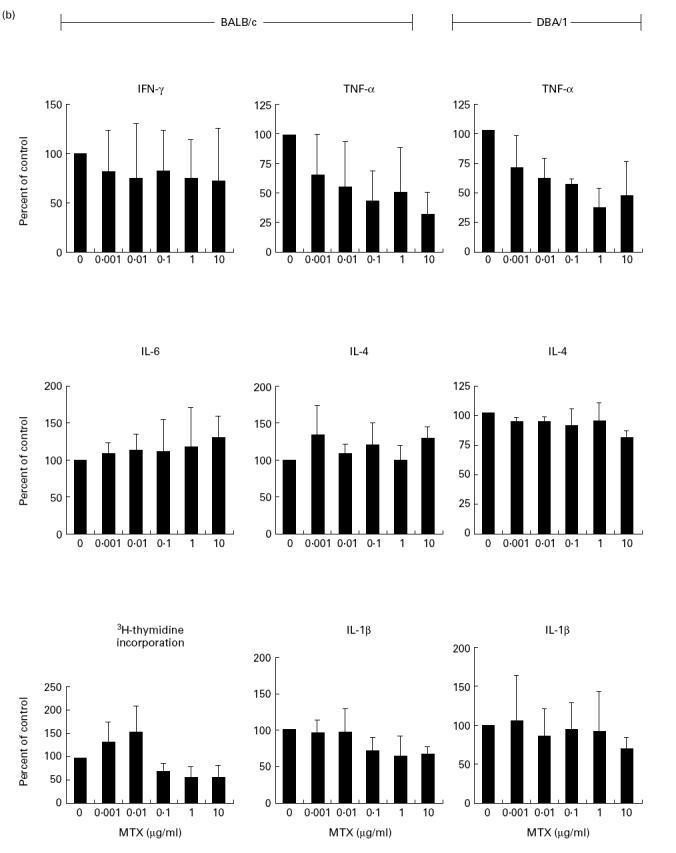
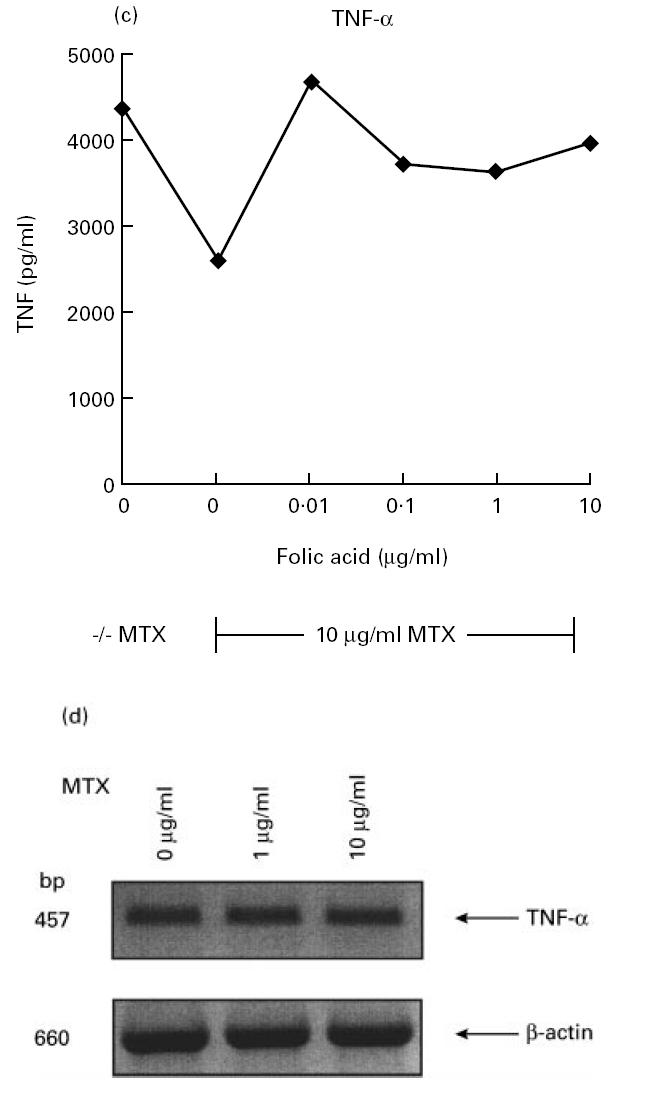
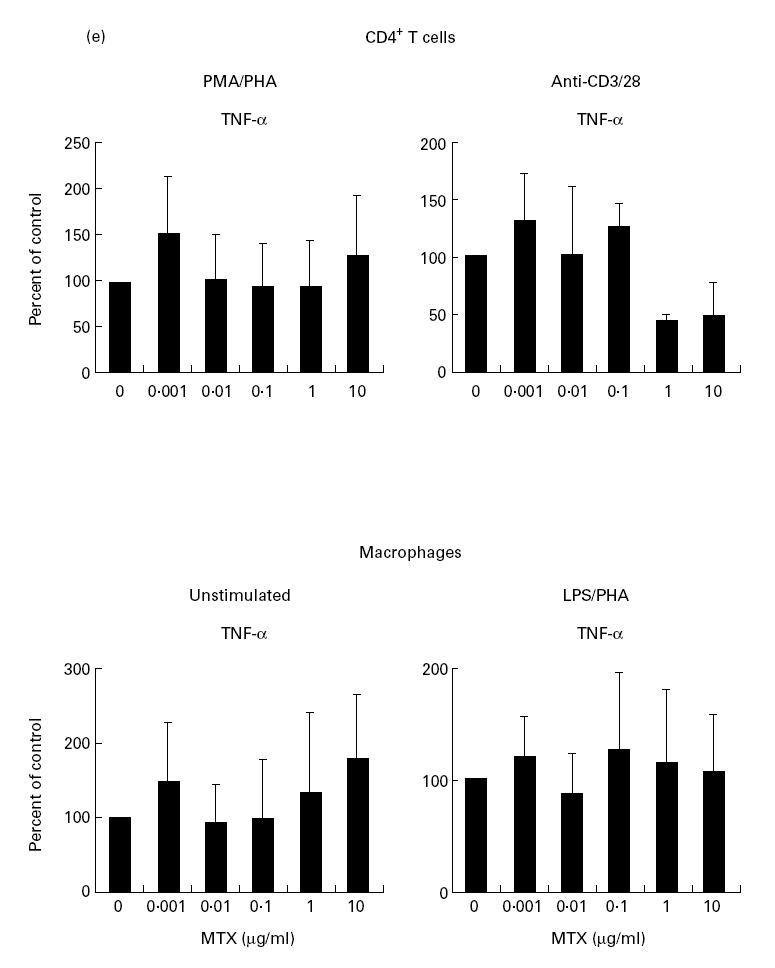
(a) Time course of tumour necrosis factor (TNF) production by cultured unstimulated (□) and stimulated (▪) murine spleen cells from BALB/c mice. Spleen cells were stimulated with anti-CD3/CD28, phorbol myristate acetate (PMA) and lipopolysaccharide (LPS), as specified in Materials and Methods. Culture supernatants were taken at indicated time points and cytokine concentrations were assessed by ELISA. Data represent mean values ± s.d. of three independent experiments. (b) Cytokine production and proliferation of methotrexate (MTX)-treated splenic cells. Production of cytokines by cultured murine spleen cells from BALB/c and DBA/1 mice in the presence or absence of indicated concentrations of MTX. Spleen cells were stimulated for 48 h with anti-CD3/CD28, PMA and LPS, as specified in Materials and Methods. Culture supernatants were taken after 48 h and cytokine concentrations were assessed by ELISA. Data represent mean values ± s.d. of measurements from three independent experiments compared with untreated cells (100%). Baseline levels for cytokine concentrations in BALB/c mice were as follows: IFN-γ 10 974 ± 135 pg/ml; TNF 1249 ± 156 pg/ml; IL-1 371 ± 38 pg/ml; IL-4 458 ± 58 pg/ml; IL-6 3940 ± 412 pg/ml. Proliferation assay of MTX-treated murine spleen cells. Spleen cells were stimulated for 48 h with PMA and PHA, as specified in Materials and Methods. Thymidine incorporation was assessed during the last 12 h of cell culture. Data represent mean values ± s.d. from three independent experiments compared with untreated cells (100%). (c) Effect of folic acid on MTX-induced suppression of TNF production by cultured murine spleen cells. Spleen cells were stimulated with anti-CD3/CD28, PMA and LPS in the presence or absence of 10 μg/ml MTX and indicated concentrations of folic acid. Culture supernatants were taken after 48 h and cytokine concentrations were assessed by ELISA. Cell viability was > 90% under all culture conditions. as assessed by trypan blue exclusion. (d) Analysis of TNF mRNA levels in MTX-treated spleen mononuclear cells by reverse transcriptase-polymerase chain reaction (RT-PCR). Spleen cells were stimulated with anti-CD3/CD28, PMA and LPS in the presence of indicated dosages of MTX. Total RNA was isolated after 48 h, transcribed into cDNA and analysed by RT-PCR using specific primers for murine TNF and β-actin. PCR products were analysed by agarose gel electrophoresis. (e) Cytokine production of MTX-treated macrophage-enriched splenic mononuclear cells and purified spleen CD4+ T lymphocytes. Production of TNF by cultured murine spleen cells from BALB/c mice was assessed in the presence or absence of indicated concentrations of MTX. Cells were stimulated for 48 h with anti-CD3/CD28, PMA and LPS, as indicated. Culture supernatants were taken after 48 h and cytokine concentrations were assessed by ELISA. Data represent mean values ± s.d. of three independent experiments compared with untreated cells (100%). (f) IL-15-induced cytokine production and proliferation of cultured murine spleen cells in the presence or absence of indicated concentrations of MTX. Spleen cells were stimulated for 48 h with 100 ng/ml rIL-15, anti-CD3/CD28, PMA and LPS, as specified in Materials and Methods. Culture supernatants were taken after 48 h and cytokine concentrations were assessed by ELISA. Data represent mean values ± s.d. from three independent experiments compared with untreated cells (100%). Baseline levels for cytokine concentrations were as follows: IFN-γ, 20 119 ± 239 pg/ml; TNF, 5123 ± 438 pg/ml; IL-1, 406 ± 32 pg/ml; IL-4, 465 ± 54 pg/ml; IL-6, 5046 ± 615 pg/ml.
In further experiments we wanted to determine whether TNF production by purified T cells or macrophages is modulated by MTX. Accordingly, we separated CD4+ T cells from murine spleen using immunomagnetic beads and found that MTX significantly (1–10 μg/ml; P < 0.01) suppressed anti-CD3/CD28-induced TNF production rather than PMA/PHA-induced TNF production (Fig. 1e). Furthermore, MTX had virtually no effect on TNF production by macrophage-enriched spleen cells (Fig. 1e), suggesting that in this experimental system TNF production by T cells rather than macrophages is the primary target of MTX.
IL-15-induced TNF-a production is suppressed by MTX
IL-15 is a recently discovered cytokine that has been implicated in the pathogenesis of RA by inducing TNF production [28,29]. Since the above data suggest an effective suppression of TNF production by MTX, we wanted to determine in subsequent studies whether MTX was also able to suppress IL-15-induced TNF production. Accordingly, we analysed MTX effects on IL-15-stimulated splenic mononuclear cells. As shown in Fig. 1f, MTX suppressed IL-15-induced TNF production, suggesting that both induced and IL-15-augmented TNF production are modulated by MTX.
Short-term treatment of healthy mice with MTX suppresses TNF but not IL-6 production by splenic mononuclear cells
In subsequent studies, we analysed whether MTX treatment of healthy mice influences cytokine production by splenic mononuclear cells. Treatment of healthy mice by two injections of MTX (day 0, day 2) caused striking changes of proinflammatory cytokine production by mononuclear splenic cells. As shown in Fig. 2, mononuclear splenic cells isolated from MTX-treated (10–100 mg/kg) mice at day 3 produced significantly (P < 0.01) lower TNF levels than cells from PBS-treated control mice. The effect of MTX on cytokine production was further underlined by the finding that serum levels of TNF were significantly (P < 0.01) reduced in MTX-treated (10–100 mg/kg) mice (untreated, 663 ± 71 pg/ml; 10 mg/kg, 245 ± 58 pg/ml; 100 mg/kg, 142 ± 15 pg/ml). As observed in the in vitro studies described above, IL-4 and IL-6 production by spleen mononuclear cells was not affected by MTX (Fig. 2).
Fig. 2.
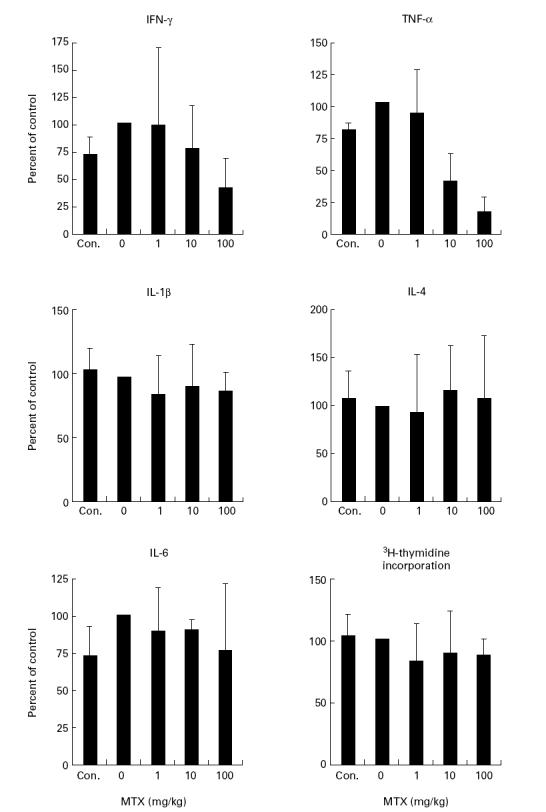
Cytokine production and proliferation of cultured murine spleen cells from methotrexate (MTX)-treated mice. BALB/c mice were treated twice (day 0, day 2) with indicated amounts of MTX, as specified in Materials and Methods. Spleen cells were isolated after 3 days and stimulated for 48 h with anti-CD3/CD28, phorbol myristate acetate (PMA) and lipopolysaccharide (LPS). Culture supernatants were taken after 48 h and cytokine concentrations were assessed by ELISA. Data represent mean values ± s.d. from three independent experiments compared with untreated cells (100%).
Long-term treatment of healthy mice with MTX suppresses TNF but not IL-4 and IL-6 production by splenic mononuclear cells ex vivo
In an additional series of studies, we analysed long-term effects of MTX on cytokine production by splenic mononuclear cells ex vivo. In these studies, we treated healthy BALB/c mice by three weekly injections of MTX for 30 days. Then we isolated splenic mononuclear cells from these mice and determined production of various pro- and anti-inflammatory cytokines (Fig. 3). While production of TNF by spleen cells was significantly (P < 0.01) suppressed upon MTX treatment, production of the TH2 type cytokine IL-4 was not strikingly reduced after long-term MTX treatment (0.01–10 mg/kg). However, in contrast to the short-term experiments described above, long-term treatment of healthy mice with MTX strongly suppressed TNF production by splenic mononuclear cells also at lower dosages (0.01–0.1 mg/kg), indicating that MTX may modulate cytokine production in a time-dependent manner.
Fig. 3.
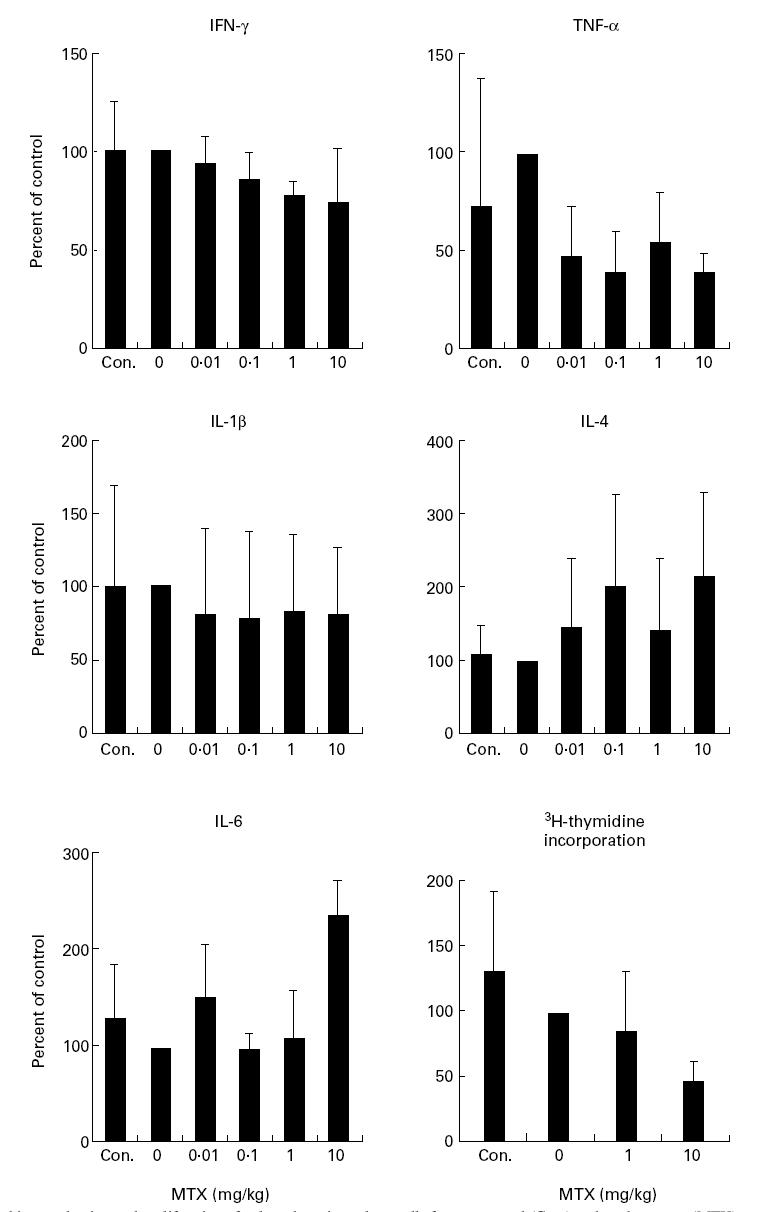
Cytokine production and proliferation of cultured murine spleen cells from untreated (Con.) and methotrexate (MTX)-treated mice. BALB/c mice were treated three times a week with indicated amounts of MTX. Spleen cells were isolated after 30 days and stimulated for 48 h with anti-CD3/CD28, phorbol myristate acetate (PMA) and lipopolysaccharide (LPS). Culture supernatants were taken after 48 h and cytokine concentrations were assessed by ELISA. Data represent mean values ± s.d. from three independent experiments compared with untreated cells (100%). Proliferation assay of cultured murine spleen cells from MTX-treated mice. BALB/c mice were treated three times a week with indicated amounts of MTX. Spleen cells were isolated after 30 days and thymidine incorporation was assessed during the last 12 h of cell culture. Data represent mean values of triplicate measurements.
MTX treatment prevents CIA in mice
The above studies indicate that MTX can modulate cytokine production by splenic mononuclear cells in vitro and in vivo. In further studies, we wanted to determine whether MTX can modulate cytokine production and clinical parameters of disease in a murine model of experimental arthritis induced by CII. When treatment with MTX was started 21 days after immunization with collagen a clear improvement both of the clinical score and of histological parameters of arthritis was observed at all dosages between 0.1 mg/kg and 10 mg/kg (Fig. 4a; Table 1). Higher doses (>1 mg/kg) of MTX completely prevented development of clinical signs of arthritis (Fig. 4a). This finding was underlined by grading of histological joint sections that demonstrated reduction of inflammatory cells and cartilage destruction in MTX-treated mice compared with the untreated control group (Table 1). In addition, isolated splenic mononuclear cells from MTX-treated mice produced less TNF and IFN-γ in cell culture (Fig. 4b). Production of IL-1β, IL-4 and IL-6, however, was unaffected by MTX treatment.
Fig. 4.
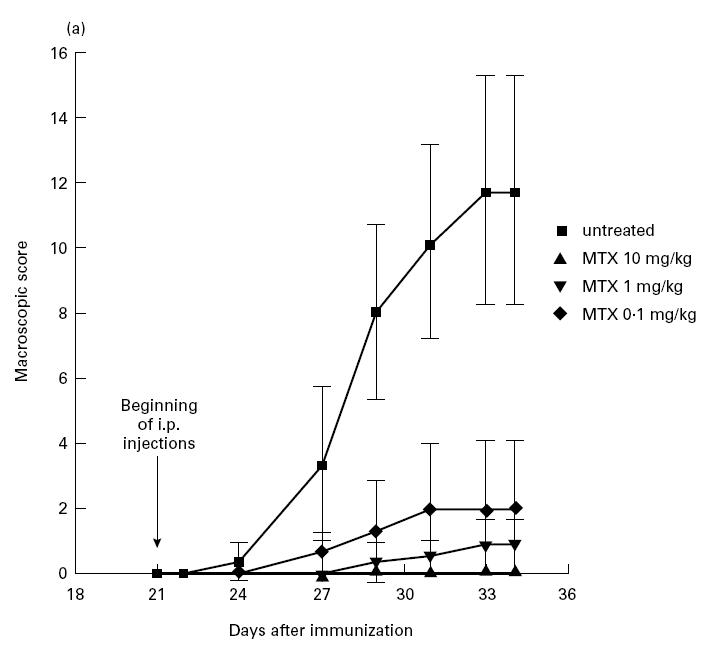
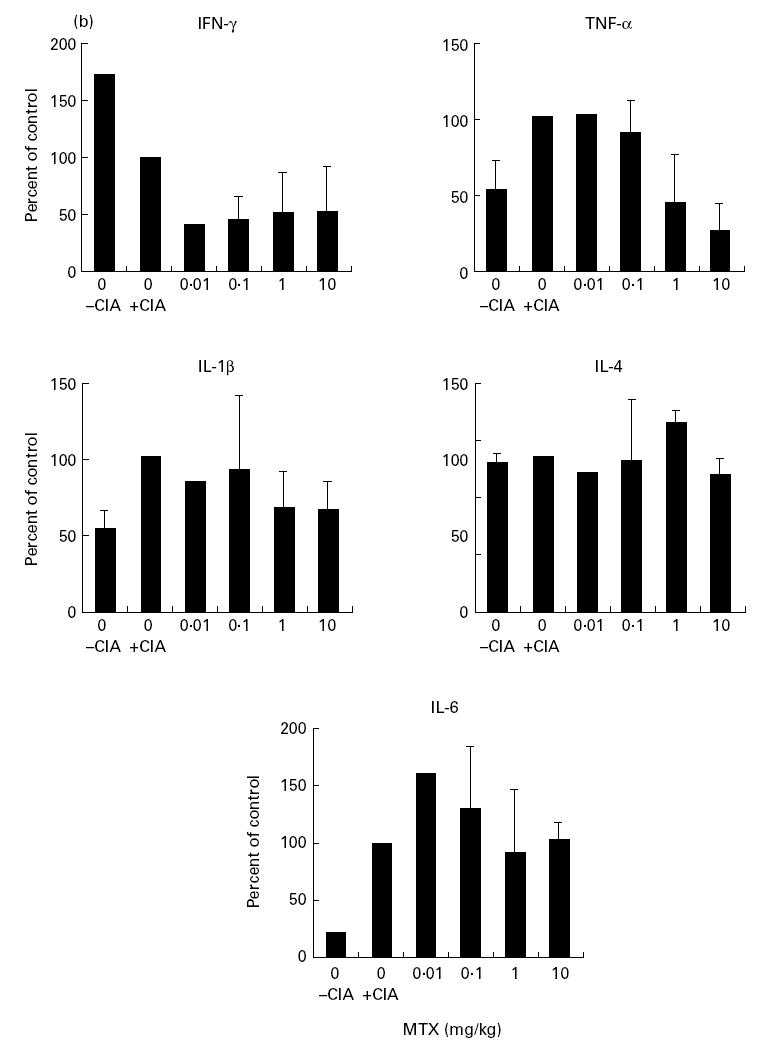
(See next page.) (a) Suppression of CIA development in mice after early treatment with methotrexate (MTX). DBA/1 mice were given indicated amounts of MTX three times a week. Treatment was started 21 days after immunization with type II collagen. Data represent mean values and s.d. of one representative experiment with at least three mice per group. Three additional independent experiments gave very similar results. (b) Production of IL-1β, IL-4, IL-6, tumour necrosis factor (TNF) and IFN-γ by cultured murine spleen cells from MTX-treated DBA/1 mice with CIA. Spleen cells were isolated 34 days after immunization and stimulated for 48 h with anti-CD3/CD28, phorbol myristate acetate (PMA) and lipopolysaccharide (LPS). Culture supernatants were taken after 48 h and cytokine concentrations were assessed by ELISA. Data represent mean values ± s.d. of three independent experiments compared with untreated cells (100%).
Table 1.
Histology after methotrexate (MTX) treatment of CIA in DBA/1 mice

Effects of MTX on cytokine production in TNF-α knockout mice and TNF-α transgenic mice
To characterize further the functional role of TNF-α in MTX-mediated effects on cytokine production, we next tried to analyse MTX effects in mice in which the gene for TNF-α had been inactivated by homologous recombination [23]. It was found that TNF−/− mice showed no significant reduction in production of IL-6 (not shown) and IFN-γ (Fig. 5) by splenic cells after long-term MTX treatment. Finally, we chose to analyse MTX effects in Tg197 mice that express in their tissues a human TNF-α transgene, as previously described [12]. When TNF transgenic Tg197 mice were treated with MTX, we observed a reduction or complete absence of arthritis activity in MTX-treated animals compared with controls. TNF production in TNF transgenic mice was again suppressed upon MTX treatment (human TNF, 37 ng/ml versus 0 ng/ml; murine TNF, 582 pg/ml versus 203 pg/ml). Furthermore, changes in IFN-γ production were more pronounced compared with TNF knockout mice, suggesting that MTX effects on IFN-γ production may be secondary to its effects on TNF production.
Fig. 5.
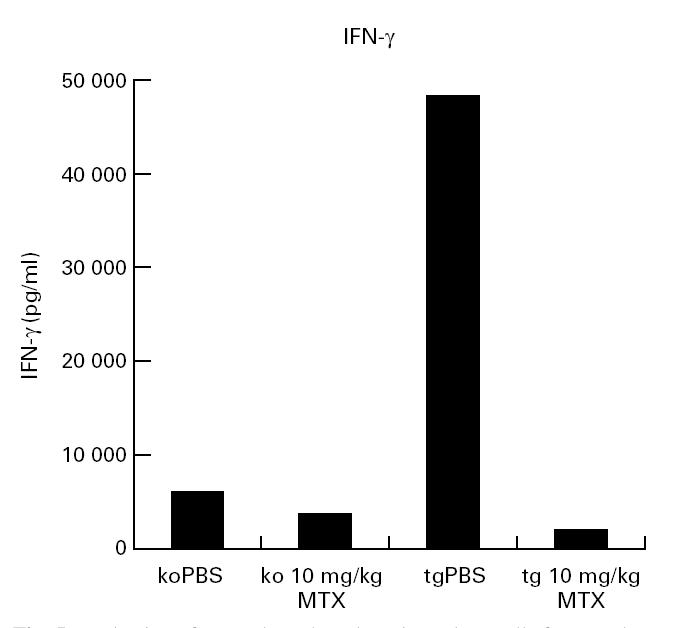
Production of IFN-γ by cultured murine spleen cells from methotrexate (MTX)-treated mice. Tumour necrosis factor (TNF) knockout(ko) and TNF transgenic (tg) mice were treated three times a week with PBS or indicated amounts of MTX for a period of 30 days. Spleen cells were isolated after 30 days and stimulated for 48 h with anti-CD3/CD28, phorbol myristate acetate (PMA) and lipopolysaccharide (LPS). Culture supernatants were taken after 48 h and cytokine concentrations were assessed by ELISA. Data represent mean values of duplicate measurements from one representative experiment. IFN-γ baseline level in uninjected transgenic mice was 43 000 pg/ml.
DISCUSSION
In the present study, we have found specific modulation of T cell and macrophage cytokine production by methotrexate (MTX), an immunosuppressive drug frequently used for treatment of patients with RA [5,15,16,30,31]. First, we demonstrated a significant suppression of TNF-α production by spleen cells after MTX treatment; second, MTX treatment of mice caused strikingly reduced TNF serum levels in vivo and diminished TNF production by spleen cells ex vivo. Third, MTX suppressed TNF production in a murine model of experimental arthritis; and fourth, we showed that MTX effects on cytokine production were reduced in TNF knockout mice but augmented in TNF-α transgenic mice. Taken together with the increased TNF-α levels in patients with RA in humans, these data suggest that suppression of production of proinflammatory cytokines could be an important target mechanism for MTX effects.
Although inhibition of B cell, T cell and macrophage activity has been suggested as important mechanism of MTX function, its mechanism of action has not been completely delineated. In particular, only few studies have analysed its effects on cytokine production in mice and humans [21,22,32,33]. These studies showed diminished IL-1 concentrations in the synovial fluid of MTX-treated patients, whereas IL-1 serum levels were not affected [21,32]. Furthermore, MTX treatment decreased TNF concentrations in the synovial fluid in experimental adjuvant arthritis, and treatment with liposomal MTX reduced TNF production ex vivo in this model [34,35]. However, no significant effect on TNF concentration in the peripheral blood of MTX-treated patients has been observed [33,36,37]. This lack of decreased TNF serum levels could be due to the fact that MTX concentrations in the synovium and bone of RA patients are known to be roughly 10-fold higher compared with plasma levels [38]. Thus, low-dose treatment with MTX in RA could be capable of suppressing local TNF production in the joint, whereas systemic TNF levels would not be affected.
With regard to the mechanism of TNF suppression by MTX it was found that MTX suppresses TNF production by anti-CD3/CD28 activated purified T cells rather than TNF secretion by macrophages, suggesting that activation of TNF production via the T cell receptor might be an important target for the action of MTX. However, suppression of TNF production by MTX in purified T lymphocytes was significantly lower (0.1–10 μg/ml; P < 0.01) and occurred at higher dosages compared with total spleen mononuclear cells, giving rise to the possibility that MTX may also modulate TNF production that depends on cell–cell interactions between splenic mononuclear cells, such as between macrophages and T cells. This finding may thus be relevant for the situation in human RA, since macrophages are the main producers of TNF in RA joints [1–3]. Furthermore, the fact that MTX effects on TNF production could be partially counteracted by folic acid suggests that the effect of MTX on TNF production is at least partially folate-dependent. In any case, the data clearly show that MTX can significantly suppress TNF production by spleen mononuclear cells. In particular, it was found that long-term treatment of mice with MTX dosages as low as 0.004 mg/kg per day leads to suppression of TNF production by spleen cells. Although there are limited data on comparative MTX drug kinetics between mice and humans, these data suggest that dosages used in our murine experiments could be relevant to those used for treatment of RA patients (e.g. 15 mg MTX for a 70-kg RA patient = 0.03 mg/kg per day) [1,31,33].
In additional studies, we analysed the effects of MTX on production of various other pro- and anti-inflammatory cytokines produced by macrophages and T cells. We found that MTX suppresses production of proinflammatory cytokines such as IL-6, IL-1β, TNF and IFN-γin vitro, ex vivo and in vivo, while production of the anti-inflammatory TH2 cytokine IL-4 was less affected. Furthermore, IL-6 production ex vivo was only suppressed after long-term treatment of mice with MTX. This characteristic dose- and time-dependent modulation of cytokine profiles by MTX argues against a general inhibitory effect on mRNA and protein synthesis. Furthermore, the observed effects on cytokine production occurred already at concentrations of MTX that did not affect cell proliferation. Thus, it is rather likely that MTX has additional specific effects on cytokine production by T cells and macrophages, leading to a clearly time-dependent selective suppression in the production of some cytokines. The suppression of proinflammatory cytokines such as TNF and IFN-γ could be important for clinical effects of MTX, since an increased production of these cytokines and a shift towards a TH1 T cell phenotype have been described in patients with RA [39]. In support of this theory, Constantin et al. [40] have recently described suppression of Thl cytokine levels in peripheral blood mononuclear cells (PBMC) from MTX-treated patients. Furthermore, we have recently found that MTX strikingly suppresses IFN-γ and TNF protein production by cultured human CD45RA and CD45RO CD4+ T lymphocytes (Hildner et al., manuscript in preparation). In our murine experimental system, however, the effects of MTX on TNF production by spleen cells were more strikingly compared with its effects on IFN-γ production. Furthermore, the effect of MTX on production of the latter cytokine was augmented in TNF transgenic Tg197 mice. In contrast, MTX effects on IFN-γ production were reduced in TNF knockout mice, suggesting that at least in our experimental system this effect may be secondary to MTX effects on TNF production.
CIA is a widely used animal model of experimental polyarthritis [41,42]. The murine CIA model of experimental arthritis contains several features that are consistent with those observed in RA in humans [39–42]: (i) a chronic arthritis is induced that is characterized by a severe synovial inflammation; (ii) there are similarities at the histological level with synovial infiltrates of macrophages and T cells leading to cartilage destruction; (iii) there is an increased production of the proinflammatory cytokine TNF-α in CIA consistent with the increased TNF-α levels previously reported in patients with RA [1,2]. Here, we demonstrate that MTX treatment completely prevents CIA, although it should be noted that MTX was administered prior to the onset of clinical arthritis, a situation which is different from treating established disease. This effect of MTX was accompanied by characteristic changes of cytokine profiles, in particular a reduction of TNF levels. Taken together, these data suggest that the observed effect on clinical parameters of CIA by MTX could be at least partially due to its effect on TNF production. This reduction of TNF levels would then lead to prevention of CIA.
The data reported here provide direct evidence for a regulatory effect of MTX on cytokine production in mice in vitro, in vivo, and ex vivo. In particular, MTX specifically modulates TNF-α production and production of this cytokine by T cells rather than macrophages appears to be the primary target of MTX. Furthermore, MTX treatment prevents experimental murine CIA. These data may serve as a basis to understand and further analyse MTX-mediated mechanisms of immunosuppression in patients with RA.
Acknowledgments
The work of M.F.N. was kindly supported by the Deutsche Forschungsgemeinschaft (DFG), the Gerhard-Hess program of the DFG (Ne 490/1-1, Ne 49012-1, Ne 490/3-1) and by the Innovationsstiftung Rheinland-Pfalz. T.G. and E.M.-H. are supported by DFG (SFB 311 ‘Immunpathogenese’). P.S. was supported by the Innovationsstiftung Rheinland-Pfalz.
REFERENCES
- 1.Kavanaugh AF, Lipsky PE. Rheumatoid arthritis. In: Rich RR, editor. Clinical immunology. St Louis: Mosby; 1996. pp. 1093–116. [Google Scholar]
- 2.Müller-Ladner U. Molecular and cellular interactions in rheumatoid synovium. Curr Opin Rheumatol. 1996;8:210–20. doi: 10.1097/00002281-199605000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Firestein GS, Alvaro-Garcia JM, Maki R. Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol. 1990;144:3347–53. [PubMed] [Google Scholar]
- 4.Chen E, Keystone EC, Fish EN. Restricted cytokine expression in rheumatoid arthritis. Arthritis Rheum. 1993;36:901–10. doi: 10.1002/art.1780360706. [DOI] [PubMed] [Google Scholar]
- 5.Moreland LW, Heck LW, Jr, Koopman WJ. Biologic agents for treating rheumatoid arthritis: concepts and progress. Arthritis Rheum. 1997;40:397–409. doi: 10.1002/art.1780400302. [DOI] [PubMed] [Google Scholar]
- 6.Elliott MJ, Maini RN, Feldmann M, et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 1993;36:1681–90. doi: 10.1002/art.1780361206. [DOI] [PubMed] [Google Scholar]
- 7.Elliott MJ, Maini RN, Feldmann M, et al. Randomised double- blind comparison of chimeric monoclonal antibody to tumor necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–10. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 8.Beutler B, Cerami A. The biology of cachectin/TNF—a primary mediator of the host response. Ann Rev Immunol. 1989;7:625–55. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 9.Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–3. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 10.Strober W, Kelsall BL, Fuss I, Marth T, Ludviksson B, Ehrhardt R, Neurath MF. Reciprocal IFN-γ and TGF-β responses regulate the occurrence of mucosal inflammation. Immunol Today. 1997;18:61–64. doi: 10.1016/s0167-5699(97)01000-1. [DOI] [PubMed] [Google Scholar]
- 11.Neurath MF, Pettersson S, Meyer zurn Büschenfelde KH, Strober W. Local administration of antisense oligonucleotides to the p65 subunit of NF-kappaB abrogates experimental colitis in mice. Nature Med. 1996;2:998–1004. doi: 10.1038/nm0996-998. [DOI] [PubMed] [Google Scholar]
- 12.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis: a predictive genetic model of arthritis. EMBO. 1991;10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon JR, Galli SJ. Mast cells as a source of both preformed and immunologically inducible TNF-alpha/cachectin. Nature. 1990;346:274–6. doi: 10.1038/346274a0. [DOI] [PubMed] [Google Scholar]
- 14.Brennan FM, Maini RN, Feldmann M. TNF-alpha: a pivotal role in rheumatoid arthritis. Br J Rheumatol. 1993;31:293–8. doi: 10.1093/rheumatology/31.5.293. [DOI] [PubMed] [Google Scholar]
- 15.Cronstein BN. Molecular therapeutics: methotrexate and its mechanism of action. Arthritis Rheum. 1996;39:1951–60. doi: 10.1002/art.1780391203. [DOI] [PubMed] [Google Scholar]
- 16.Weinblatt ME, Kaplen H, Germain BF, et al. Methotrexate in rheumatoid arthritis: a five year prospective multicenter study. Arthritis Rheum. 1994;37:1492–8. doi: 10.1002/art.1780371013. [DOI] [PubMed] [Google Scholar]
- 17.Hu SK, Mitcho YL, Oronsky AL, Kerwar SS. Studies on the effect of methotrexate on macrophage function. J Rheumatol. 1988;15:206–9. [PubMed] [Google Scholar]
- 18.Conolly KM, Stecher VJ, Danis E, Pruden DJ, LaBrie T. Alteration of interleukin-1 production and the acute phase response following medication of adjuvant arthritic rats with cyclosporin-A or methotrexate. Int J Immunopharmacol. 1988;10:717–28. doi: 10.1016/0192-0561(88)90025-2. [DOI] [PubMed] [Google Scholar]
- 19.Segal R, Mozes E, Yaron M, Tartakovsky B. The effects of methotrexate on the production and activity of interleukin-l. Arthritis Rheum. 1989;32:370–7. doi: 10.1002/anr.1780320403. [DOI] [PubMed] [Google Scholar]
- 20.Meyer FA, Yaron I, Mashiah V, Yaron M. Methotrexate inhibits proliferation but not interleukin 1 stimulated secretory activities of cultured human synovial fibroblasts. J Rheumatol. 1993;20:238–42. [PubMed] [Google Scholar]
- 21.Seitz M, Loetscher P, Dewald B, Towbin H, Rordorf C, Gallati H, Baggiolini M, Gerber NJ. Methotrexate action in rheumatoid arthritis: stimulation of cytokine inhibitor and inhibition of chemokine production by peripheral blood mononuclear cells. Br J Rheumatol. 1995;34:602–9. doi: 10.1093/rheumatology/34.7.602. [DOI] [PubMed] [Google Scholar]
- 22.Brody M, Bohm I, Bauer R. Mechanism of action of methotrexate: experimental evidence that methotrexate blocks the binding of interleukin 1-beta to the interleukin 1 receptor on target cells. Eur J Clin Chem Clin Biochem. 1993;31:667–74. doi: 10.1515/cclm.1993.31.10.667. [DOI] [PubMed] [Google Scholar]
- 23.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF-alpha-deficient mice: a critical requirement for TNF-alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rook G, Thompson S, Buckley M, Elson C, Brealey R, Lambert C, White T, Rademacher T. The role of oil and agalactosyl IgG in the induction of arthritis in rodent models. Eur J Immunol. 1991;21:1027–32. doi: 10.1002/eji.1830210425. [DOI] [PubMed] [Google Scholar]
- 25.Germann T, Szeliga J, Hess H, Storkel S, Podlaski FJ, Gately MK, Schmitt E, Rude E. Administration of interleukin-12 in combination with type 11 collagen induces severe arthritis in DBA/1 mice. Proc Natl Acad Sci USA. 1995;92:4823–7. doi: 10.1073/pnas.92.11.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joosten LAB, Lubberts E, Durez P, Helsen MMA, Jacobs MJM, Goldman M, van den Berg WB. Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis: protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum. 1997;40:249–60. doi: 10.1002/art.1780400209. [DOI] [PubMed] [Google Scholar]
- 27.Barbulescu K, Becker C, Schlaak JF, Schmitt E, Meyer zum Büschenfelde KH, Neurath MF. Cutting edge: IL-12 and IL-18 differentially regulate the transcriptional activity of the human interferon-gamma pro-moter in primary CD4+ T lymphocytes. J Immunol. 1998;160:3642–7. [PubMed] [Google Scholar]
- 28.McInnes IB, Leung BP, Sturrock RD, Field M, Liew FY. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nature Med. 1997;3:189–95. doi: 10.1038/nm0297-189. [DOI] [PubMed] [Google Scholar]
- 29.McInnes IB, al-Mughales J, Field M, et al. The role of interleukin-15 in T-cell migration and activation in rheumatoid arthritis. Nature Med. 1996;2:175–82. doi: 10.1038/nm0296-175. [DOI] [PubMed] [Google Scholar]
- 30.Kremer JM, Phelps CT. Long-term prospective study of the use of methotrexate in the treatment of rheumatoid arthritis: update after a mean of 90 months. Arthritis Rheum. 1992;35:138–45. doi: 10.1002/art.1780350203. [DOI] [PubMed] [Google Scholar]
- 31.Weinblatt ME, Coblyn JS, Fox DA, Fraser PA, Holdsworth DE, Glass DN, Trentham DE. Efficacy of low-dose methotrexate in rheumatoid arthritis. N Engl J Med. 1985;312:88–95. doi: 10.1056/NEJM198503283121303. [DOI] [PubMed] [Google Scholar]
- 32.Thomas R, Carroll GJ. Reduction of leucocyte and interleukin-1-beta concentrations in the synovial fluid of rheumatoid arthritis patients treated with methotrexate. Arthritis Rheum. 1993;36:1244–52. doi: 10.1002/art.1780360909. [DOI] [PubMed] [Google Scholar]
- 33.Barrera BR, Haagsma CJ, Boerbooms AM, Van Riel PL, Borm GF, Van dePutte LB, Van der Meer JW. Effect of methotrexate alone or in combination with sulphasalazine on the production and circulating concentrations of cytokines and their antagonists. Longitudinal evaluation in patients with rheumatoid arthritis. Br J Rheumatol. 1995;34:747–55. doi: 10.1093/rheumatology/34.8.747. [DOI] [PubMed] [Google Scholar]
- 34.Smith-Oliver T, Noel LS, Stimpson SS, Yarnall DP, Conolly KM. Elevated levels of TNF in the joints of adjuvant arthritic rats. Cytokine. 1993;5:298–304. doi: 10.1016/1043-4666(93)90060-i. [DOI] [PubMed] [Google Scholar]
- 35.Williams AS, Camilleri JP, Topley N, Williams BD. Prostaglandin and tumor necrosis factor secretion by peritoneal macrophages isolated from normal and arthritic rats treated with liposomal methotrexate. J Pharmacol Toxicol Methods. 1994;32:53–58. doi: 10.1016/1056-8719(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 36.Chang DM, Weinblatt ME, Schur PH. The effects of methotrexate on interleukin-1 in patients with rheumatoid arthritis. J Rheumatol. 1992;19:1678–82. [PubMed] [Google Scholar]
- 37.Williams AS, Punn YL, Amos N, Cooper AM, Williams BD. The effect of liposomally conjugated methotrexate upon mediator release from human peripheral blood monocytes. Br J Rheumatol. 1995;34:241–5. doi: 10.1093/rheumatology/34.3.241. [DOI] [PubMed] [Google Scholar]
- 38.Bologna C, Edno L, Anaya JM, et al. Methotrexate concentrations in synovial membrane and trabecular and cortical bone in rheumatoid arthritis patients. Arthritis Rheum. 1994;37:1770–3. doi: 10.1002/art.1780371210. [DOI] [PubMed] [Google Scholar]
- 39.Dolhain RJEM, van der Heiden AN, ter Haar NT, Breedveld FC, Miltenburg AMM. Shift toward T lymphocytes with a T helper 1 cytokine-secretion profile in the joints of patients with rheumatoid arthritis. Arthritis Rheum. 1996;39:1961–9. doi: 10.1002/art.1780391204. [DOI] [PubMed] [Google Scholar]
- 40.Constantin A, Loubet P, Lambert N, Yassine B, Abbal M, Mazieres B, Preval C, Cantagrel A. Antiinflammatory and immunoregulatory action of methotrexate in the treatment of rheumatoid arthritis. Arthritis Rheum. 1998;41:48–57. doi: 10.1002/1529-0131(199801)41:1<48::AID-ART7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 41.Trentharn DE, Townes AS, Kang AH. Autoimmunity to type II collagen: an experimental model of arthritis. J Exp Med. 1977;146:857–68. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joints disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992;89:9784–8. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]