

Abstract
As Q fever is associated with an inflammatory syndrome, we determined circulating levels of inflammatory cytokines, cytokine antagonists, and activation markers of leucocytes in patients with acute Q fever and Q fever endocarditis. Tumour necrosis factor (TNF) and IL-6, but not IL-1β, were markedly increased compared with controls. Cytokine antagonists and activation markers of leucocytes were profoundly different in acute and chronic Q fever. IL-1 receptor antagonist and TNF receptor type II were significantly increased in patients with acute Q fever, suggesting a shift of cytokine balance towards cytokine antagonists. The activation marker of B cells, sCD23, was significantly increased in Q fever endocarditis compared with controls and patients with acute Q fever. In a 2-year follow-up study of patients with Q fever endocarditis, sCD23 and specific IgG levels slowly decreased in patients whose symptoms resolved, but remained high in those who required prolonged treatment.
Keywords: Q fever, inflammatory cytokines, cytokine antagonists, leucocyte markers, sCD23
INTRODUCTION
Q fever, caused by Coxiella burnetii, is commonly divided into acute and chronic forms. Acute Q fever manifestations usually include a self-limited flu-like syndrome, atypical pneumonia and granulomatous hepatitis [1]. The major form of chronic Q fever is endocarditis, which occurs in patients with valvular damage and/or immune dysfunction. Q fever endocarditis is associated with an inflammatory syndrome manifesting as anaemia, increased erythrocyte sedimentation rate and polyclonal hypergammaglobulinaemia [2].
Immune response plays an essential role in C. burnetii infections [3]. Acute infections are associated with an inflammatory response [4] and a protective immune response indicated by the presence of granuloma. In contrast, chronic Q fever seems to result from an inefficient response to C. burnetii, as demonstrated by the lack of granulomas, the failure of C. burnetii to induce lymphoproliferation and interferon-gamma (IFN-γ) production and the detection of high levels of antibodies to C. burnetii [1,5]. In addition, inflammatory cytokines, tumour necrosis factor (TNF) and IL-1β, are produced in excess by patient monocytes [6]. The significance of inflammatory response in the two forms of Q fever, as well as the contribution of immune cell activation, are still ignored.
This study was undertaken to investigate the balance between inflammatory cytokines (TNF, IL-1β, IL-6) and their natural inhibitors TNF receptors type I (TNF-RI) and type II (TNF-RII), or IL-1 receptor antagonist (IL-1Ra) in the plasma of patients with acute and chronic Q fever. Leucocyte activation was studied by measuring circulating markers of monocytes (neopterin, sCD14), T cells (sCD25) and B cells (sCD23). We found that IL-1Ra was up-regulated in patients with acute Q fever, while sCD23 was specifically increased in Q fever endocarditis.
PATIENTS AND METHODS
Patients
The study included 13 patients with acute Q fever, consisting of eight men and five women (mean age 33 years; range 18–48 years) and 23 patients with Q fever endocarditis, consisting of 14 men and nine women (mean age 46 years; range 20–67 years). Healthy subjects were included as controls. They consisted of 10 men and seven women (mean age 35 years; range 22–50 years). Informed consent was obtained from all patients before clinical observation and blood collection, and the study was approved by the Ethics Committee of the Université de la Méditerranée (Marseille, France). Acute Q fever was diagnosed by detection of IgM (mean titre 200; range 50–800) and IgG (mean titre 1200; range 100–3200) specific for C. burnetii in phase II. All patients were in the early phase (between 1 and 3 months after onset) of illness as demonstrated by the high levels of specific IgM. The diagnosis of Q fever endocarditis was based on modified Duke's University criteria [7] and the presence of specific IgG [1]: phase I C. burnetii IgG titres, mean 12 000, range 800–100 000; phase II C. burnetii IgG titres, mean 15 000, range 800–200 000.
Determination of cytokines and cytokine antagonists
Blood was drawn into EDTA anticoagulant tubes and plasma collected at −80°C within 2 h of collection. In order to assess potential endotoxin contamination of plasma, we used an assay based on the ability of plasma to stimulate TNF secretion in normal peripheral blood mononuclear leucocytes in the presence or the absence of polymyxin B [8]. TNF-stimulating endotoxins were not detected in patients' or controls' plasma. Circulating cytokines were measured by sandwich enzyme immunoassay (EIA). The assays were run in duplicate and were performed in accordance with the manufacturer's instructions. TNF and IL-6 were assayed using kits provided by Immunotech (Marseille, France). Their detection limit was 5 pg/ml and 3 pg/ml, respectively. IL-1β was determined using Titerzyme EIA kit (PerSeptive Diagnostics, BioAdvance, Emerainville, France). The IL-1β detection limit was about 5 pg/ml. TNF-RI, TNF-RII and IL-1Ra detection kits were from R&D Systems (Abingdon, UK). The sensitivity of the assays was 30, 10, and 20 pg/ml, respectively. The intra- and interassay coefficients of variation of EIA kits ranged between 5% and 10%.
Determination of leucocyte markers
Soluble CD14 was measured using an EIA kit manufactured by IBL (BioAdvance). Its sensitivity was 1 ng/ml. Soluble CD23 and sCD25 were assayed by sCD23 and Cell Free Interleukin 2 receptor EIA Test kits (T Cell Diagnostics, BioAdvance). The detection limits of sCD23 and sCD25 were 1.3 and 24 U/ml, respectively. Neopterin assay was performed by competitive EIA (IBL) with a sensitivity of about 0.2 ng/ml. The intra- and interassay coefficients of variation were < 10%.
Statistical analysis
Data were analysed with variance analysis (anova). Differences were considered significant when P < 0.05.
RESULTS
Inflammatory cytokines and their inhibitors in Q fever
TNF was barely detectable in 10 of 17 controls and increased significantly in all patients with Q fever (Table 1). IL-6 was detectable in three of 17 controls, in 10 of 13 patients with acute Q fever and 17 of 23 patients with endocarditis. IL-6 increased significantly in each patient group. The levels of IL-1β were low and equivalent in three subject groups. This was not the result of the presence of soluble receptors in the plasma since the recovery of recombinant human IL-1β added to plasma was about 85–95% in controls and each patient group. TNF-RI was detected to a similar extent in the three subject groups (Table 1). In contrast, TNF-RII increased markedly in all patients. IL-1Ra was detectable in 13 of 17 controls and in all patients. IL-1Ra levels were up-regulated significantly in patients with acute Q fever compared with healthy controls and patients with Q fever endocarditis.
Table 1.
Inflammatory markers of leucocytes in Q fever

Activation leucocyte markers in Q fever
Monocyte activation was evaluated using neopterin and sCD14 (Table 1). Their levels were similar in the three subject groups. T cell activation was determined by the measurement of sCD25. Soluble CD25 was detected in normal individuals and higher levels of sCD25 were found in patients. Soluble CD23 levels were assessed as markers of B cells. Similar levels of sCD23 were found in controls and in patients with acute Q fever. In contrast, sCD23 levels were four times greater in all patients with Q fever endocarditis. The levels of sCD23 were also followed for 24 months in 13 patients. Two distinct patterns were found according to the outcome of the disease (Fig. 1). In nine of 13 patients, clinical improvement and a regular decrease in specific IgG led to the end of treatment. In these patients, sCD23 levels started to decrease after 3 months and dropped significantly from 6 to 24 months (Fig. 1a). The lack of response to treatment in four of 13 patients, as determined by a slow decrease in specific IgG levels, resulted in the continuation of treatment. In these patients, sCD23 levels were not significantly different from the beginning to the end of follow up (Fig. 1b). After 24 months, sCD23 levels were significantly (P < 0.005) higher in patients who were still receiving treatment than in cured patients. Hence, sCD23 may be considered as an activity marker of Q fever endocarditis.
Fig. 1.
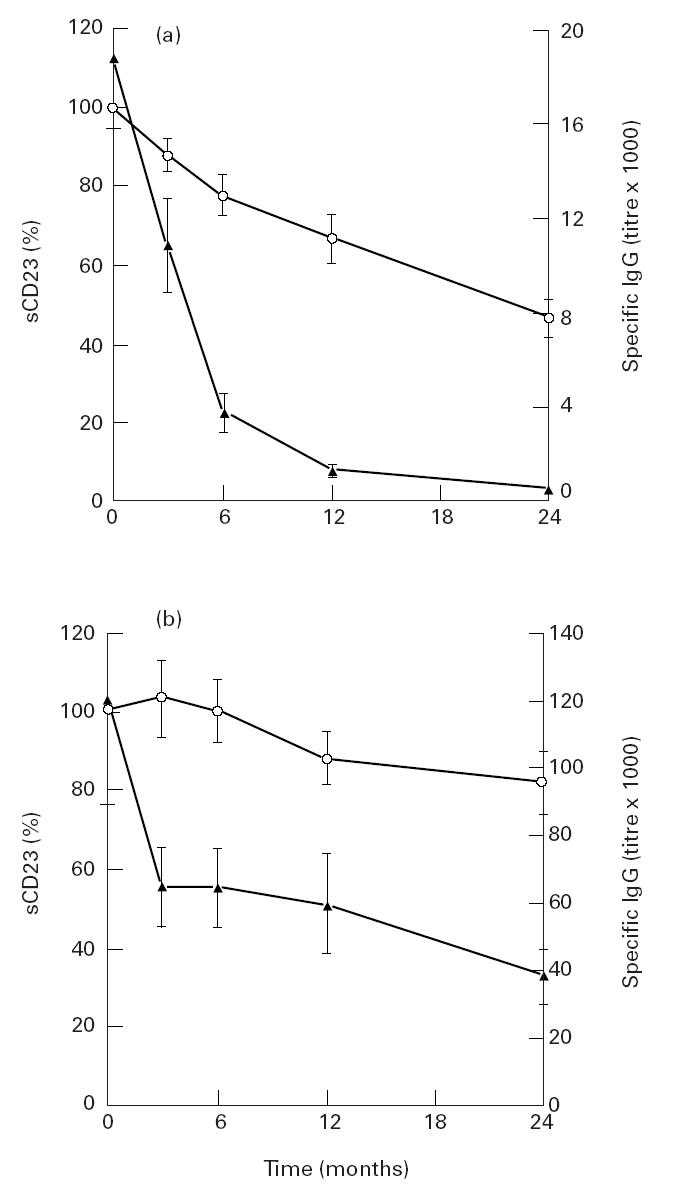
Follow up of sCD23 in Q fever endocarditis. Soluble CD23 (○) and IgG specific for phase I Coxiella burnetii (▴) were assessed in the plasma of patients with Q fever endocarditis (n = 13) every 3 months for 24 months. Two groups were determined: the first group of nine patients showed no further signs of Q fever endocarditis at the end of this period and their treatment was stopped (a, positive outcome); the second group of four patients continued to receive treatment (b, negative outcome). Results are expressed as mean ± s.e.m.
DISCUSSION
The inflammatory response in Q fever was assessed by the study of circulating cytokines. TNF and IL-6, but not IL-1β, were up-regulated in Q fever. These results extend previous findings showing that the production of TNF by monocytes is markedly enhanced in patients with Q fever endocarditis [6]. The inflammatory response to infection is often associated with the production of cytokine antagonists such as soluble receptors for cytokines. Indeed, large amounts of TNF-R are simultaneously found with TNF [9]. Although TNF-RI levels were similar in patients and controls, TNF-RII levels were enhanced in Q fever. It is likely that the increase in TNF levels results in the up-regulation of TNF-RII. Soluble TNF-R neutralize the effects of TNF and the TNF/TNF-RI ratio is of prognostic value in meningococcaemia [9]. However, sTNF-R may also stabilize TNF structure and preserve its activity, thus favouring protracted effects of TNF [10]. The circulating levels of IL-1Ra, known to block the activity of IL-1, are increased in sepsis and HIV infection. IL-1Ra is also significantly expressed in granulomatous lesions of tuberculosis [11]. Our data are the first demonstration of an IL-1Ra increase in the acute phase of an infectious disease caused by an intracellular microorganism. The lack of IL-1β and the high levels of IL-1Ra may contribute to the decreased resistance to C. burnetii infection, as described for Listeria monocytogenes in transgenic mice over-expressing IL-1Ra [11]. We cannot rule out that an IL-1Ra increase in acute Q fever is one component of the inflammatory syndrome, since IL-1Ra is now considered as an acute-phase protein. Although the role of IL-1Ra in Q fever remains speculative, its high levels in plasma may prove a useful diagnostic marker for Q fever.
The contribution of leucocyte activation markers in Q fever was then assessed. While monocyte and T cell activation markers were either not affected or non-specifically increased in C. burnetii infection, sCD23, mainly produced by B cells in response to IL-4, was specifically associated with Q fever endocarditis. Soluble CD23, the low-affinity receptor for IgE, potentiates IgE synthesis and acts as an autocrine growth factor [12]. It also triggers cytokine release in monocytes [13], suggesting a role in inflammatory responses. An sCD23 increase has been reported in parasitic and chronic inflammatory diseases such as sarcoidosis [14]. Our data extend the contribution of sCD23 to a chronic disease caused by an obligate intracellular bacterium. They are of interest for two reasons. First, the increase in sCD23 may evoke a skewing of immune response towards humoral immunity, as suggested for the hypergammaglobulinaemia of sarcoidosis [14]. In this context, elevated sCD23 levels may give some insights into the understanding of high levels of specific IgG found in Q fever endocarditis [1]. They do not, however, mean that the immune response against C. burnetii is polarized towards a Th2-type response. Indeed, we have recently shown that C. burnetii-specific antibodies belong mainly to the IgG1 subclass [15]. We have also demonstrated an overproduction of IL-10 by resting monocytes from patients with Q fever endocarditis [16], but not in response to C. burnetii antigen (unpublished data). Thus, the role of the Th2-type immune response in Q fever endocarditis is still debatable. Second, there is a relationship between the outcome of Q fever endocarditis and the levels of sCD23. The kinetics of sCD23 and specific IgG decrease were similar in patients who were successfully treated. In patients in whom treatment was continued, sCD23 levels were constant during follow up, whereas specific IgG levels slowly decreased. As the main problem of chronic Q fever treatment is the evaluation of therapeutic success [1], follow up of sCD23 may be useful.
In conclusion, this study shows the prominent role of cytokine inhibitors in the inflammatory syndrome of acute Q fever. It also demonstrates that sCD23 levels may constitute an important diagnosis marker of Q fever endocarditis and allow the evaluation of response to the treatment.
Acknowledgments
This work was supported by the Programme Hospitalier de Recherche Clinique 1996, Assistance Publique-Hôpitaux de Marseille (investigator: C.C.).
REFERENCES
- 1.Raoult D, Marrie T. Q fever. Clin Infect Dis. 1995;20:489–96. doi: 10.1093/clinids/20.3.489. [DOI] [PubMed] [Google Scholar]
- 2.Raoult D. Host factors in the severity of Q fever. Ann NY Acad Sci. 1990;590:33–38. doi: 10.1111/j.1749-6632.1990.tb42204.x. [DOI] [PubMed] [Google Scholar]
- 3.Mege JL, Maurin M, Capo C, et al. Coxiella burnetii: the ‘query’ fever bacterium. A model of immune subversion by a strictly intracellular microorganism. FEMS Microbiol Rev. 1997;19:209–17. doi: 10.1111/j.1574-6976.1997.tb00298.x. [DOI] [PubMed] [Google Scholar]
- 4.Penttila IA, Harris RJ, Storm P, et al. Cytokine dysregulation in the post-Q-fever fatigue syndrome. Q J Med. 1998;91:549–60. doi: 10.1093/qjmed/91.8.549. [DOI] [PubMed] [Google Scholar]
- 5.Izzo AA, Marmion BP. Variation in interferon-gamma responses to Coxiella burnetii antigens with lymphocytes from vaccinated and naturally infected subjects. Clin Exp Immunol. 1993;94:507–15. doi: 10.1111/j.1365-2249.1993.tb08226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capo C, Zugun F, Stein A, et al. Upregulation of tumor necrosis factor alpha and interleukin-1β in Q fever endocarditis. Infect Immun. 1996;64:1638–42. doi: 10.1128/iai.64.5.1638-1642.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fournier PE, Casalta JP, Habib G, et al. Modification of the diagnostic criteria proposed by the Duke endocarditis service to permit improved diagnosis of Q fever endocarditis. Am J Med. 1996;100:629–33. doi: 10.1016/s0002-9343(96)00040-x. [DOI] [PubMed] [Google Scholar]
- 8.Mege JL, Brunet P, Capo C, et al. Non-endotoxinic tumour necrosis factor-α-inducing factors in haemodialysis. Nephrol Dial Transplant. 1994;9:1606–10. [PubMed] [Google Scholar]
- 9.Olsson I, Gatanaga T, Gullberg U, et al. Tumour necrosis factor (TNF) binding proteins (soluble TNF receptor forms) with possible roles in inflammation and malignancy. Eur Cytokine Netw. 1993;4:169–80. [PubMed] [Google Scholar]
- 10.Aderka D, Engelmann H, Maor Y, et al. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J Exp Med. 1992;175:323–9. doi: 10.1084/jem.175.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arend NP, Malyak M, Guthridge CJ, et al. Interleukin-1 receptor antagonist: role in biology. Annu Rev Immunol. 1998;16:27–55. doi: 10.1146/annurev.immunol.16.1.27. [DOI] [PubMed] [Google Scholar]
- 12.Conrad DH. Fcε RII/CD23: the low affinity receptor for IgE. Annu Rev Immunol. 1990;8:623–45. doi: 10.1146/annurev.iy.08.040190.003203. [DOI] [PubMed] [Google Scholar]
- 13.Armant M, Rubio M, Delespesse G, et al. Soluble CD23 directly activates monocyte to contribute to the antigen-independent stimulation of resting T cells. J Immunol. 1995;155:4868–75. [PubMed] [Google Scholar]
- 14.Bansal AS, Bruce J, Hogan PG, et al. An assessment of peripheral immunity in patients with sarcoidosis using measurements of serum vitamin D3, cytokines and soluble CD23. Clin Exp Immunol. 1997;110:92–97. doi: 10.1046/j.1365-2249.1997.4921389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capo C, Iorgulescu I, Mutillod M, et al. Increases in the levels of Coxiella burnetii-specific immunoglobulins G1 and G3 antibodies in acute Q fever and chronic Q fever. Clin Diag Lab Immunol. 1998 doi: 10.1128/cdli.5.6.814-816.1998. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Capo C, Zaffran Y, Zugun F, et al. Production of interleukin-10 and transforming growth factor β by peripheral blood mononuclear cells in Q fever endocarditis. Infect Immun. 1996;64:4143–7. doi: 10.1128/iai.64.10.4143-4147.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]