

Abstract
In vitro, prostaglandins (PG) have strong inhibitory effects on T cell activation and proliferation and inhibitors of PG synthesis (NSAID) increase proliferation and activation of T cells. Although most studies have failed to demonstrate cyclooxygenase (COX) activity in lymphocytes, there is contradictory evidence on the synthesis of different PG. We have studied by reverse transcriptase-polymerase chain reaction (RT-PCR) and Western blot the expression of COX-1 and -2 mRNA and protein in resting and activated peripheral blood or Jurkat T cells. Cells were activated by T cell receptor triggering with OKT3 antibodies and activation confirmed by flow cytometric analysis of surface CD69. COX enzymatic activity was measured by determination of arachidonic acid (AA)-induced PG synthesis. Both peripheral blood and Jurkat T cells expressed COX-1 and -2 mRNA and protein. COX-1 was constitutively expressed and did not change after OKT3 stimulation. COX-2 was inducible upon OKT3-induced activation. In spite of the presence of COX mRNA and immunoreactive protein, AA-induced PG synthesis was not detected at the EIA detection (pm) level. The potential role of cyclooxygenases in T cells deserves further study, since no PG of the studied series seem to be synthesized by T cells.
Keywords: T lymphocytes, prostaglandins, prostaglandin H2 synthase
INTRODUCTION
Prostaglandin H2 synthase (cyclooxygenase or COX) is the rate-limiting enzyme in the conversion of arachidonic acid (AA) to prostaglandins (PG). PG are important mediators in physiological processes and in the inflammatory response [1]. Two COX isoenzymes have been identified. COX-1 is constitutively expressed by most cell types and it is thought to participate in PG synthesis during physiological processes [2]. Unlike COX-1, COX-2 is not constitutively expressed in normal circumstances, but it is rapidly induced by stimuli which differ according to the particular cell type [3–6]. In leucocytes and connective tissue resident cells, cytokines and other proinflammatory stimuli are the major COX-2 inducers. Specific COX-2 inhibitors that target the inflammation-associated isoenzyme while preserving the physiological synthesis of PG have been recently marketed [7].
Paradoxically, PG have also anti-inflammatory and immunosuppressive effects [8]. In vitro, treatment of T lymphocytes with PG inhibits their activation and proliferation [9–11]. Conversely, treatment with such PG synthesis inhibitors as non-steroidal anti-inflammatory drugs (NSAID) increase proliferation and activation of T cells [12,13]. Data regarding the ability of T cells to synthesize PG from AA precursors are contradictory. In lymphocyte cultures, most studies failed to detect synthesis of AA products [11,14–17], yet few found evidence for the synthesis of small amounts of different PG [12,18].
In spite of the multiple effects of NSAID on T cells, expression of COX in lymphocytes has not received attention. The molecular target of these drugs in lymphocytes remains unknown. The expression and potential functions of COX isoforms in T lymphocytes may be important, since most patients with T cell-mediated rheumatic diseases, such as rheumatoid arthritis (RA), are chronically treated with NSAID.
In the present study, we investigate whether COX-1 and -2 isoforms are expressed in T cells. In agreement with previous studies, we failed to detect AA-induced PG in these cells. We found, however, that both COX-1 and COX-2 mRNA and protein are expressed and differentially regulated in human peripheral blood T cells and a human Jurkat T cell line.
MATERIALS AND METHODS
Cell preparation
Peripheral blood mononuclear cells (PBMC) were obtained from healthy blood donors and purified by Ficoll–Hypaque density gradient centrifugation. Cells were allowed to adhere to plastic dishes in RPMI medium. Non-adherent cells were collected and the remaining monocytes were removed with CD14 antibody-coated magnetic beads (Dynabeads; Dynal, Oslo, Norway). Additional purification of peripheral blood T lymphocytes (PBL) was performed through nylon wool columns. Effective removal of monocytes and quantification of T cells after purification were evaluated by flow cytometric (FACS) analysis (EPICS Elite; Coulter, Hialeah, FL) with My4 (Coulter) or anti-CD3 MoAbs (Becton Dickinson, Mountain View, CA). PBL were < 2% My4+ and 96 ± 5% CD3+. Jurkat cells were cultured on RPMI medium with 10% fetal calf serum (FCS).
Stimulation with OKT3 antibody (Ortho Diagnostic Systems Inc, Raritan, NJ) was performed by precoating plastic dishes with antibody to a final concentration of 100 ng/ml. T cell activation was confirmed by FACS analysis of surface CD69 with FITC-labelled antibody (Coulter). Adherent mononuclear cells were used as controls for COX-1 and COX-2 expression, non-stimulated or after 10 μg/ml lipopolysaccharide (LPS) stimulation for 24 h.
Synovial fluid (SF) cells were obtained by arthrocentesis from 10 patients with RA. None of the patients had received systemic or intra-articular steroids. PBMC were also obtained from these patients and healthy individuals. SF cells were extensively washed with PBS, smeared onto silanized slides and fixed for 5 min with acetone for double immunolabelling studies.
Western blot analysis
Protein from 106 cells was extracted in ice-cold lysis buffer (500 mm Tris–HCl pH 8, 10 mm EDTA, 50 mm NaCl, 1% NP-40, 0.1% SDS, 10 μg/μl leupeptin and 1 mm PMSF). The supernatant was used for protein determination with the Bradford method (BioRad Labs, Richmond, CA). Solubilized extracts (50 μg) were electrophoresed on SDS 10% polyacrylamide gel and transferred electrophoretically to nitrocellulose filters. After blocking for 2 h with 4% non-fat dried milk in Tris-buffered saline containing 0.1% Tween-20 (TBS–T), the membranes were incubated overnight at 4°C with polyclonal rabbit anti-COX-1 (1:250) (Oxford Biomedical, Oxford, MI) or monoclonal mouse anti-COX-2 (1:750) (Cayman Chemical, Ann Arbor, MI) antibodies in 1% non-fat dried milk in TBS–T. The filters were washed, incubated for 1 h with secondary antibodies linked to peroxidase at 1:1500 dilution. Bands were visualized by the enhanced chemiluminescence detection system ECL (Amersham Corp., Arlington Heights, IL).
RNA extraction and reverse transcriptase-polymerase chain reaction analysis
Total RNA was prepared by the Ultraspec RNA isolation system (Biotecx Labs, Houston, TX). Total RNA (1 μg) was reverse transcribed into cDNA by a 1-h incubation at 37°C with 200 U of Moloney Leukaemia reverse transcriptase (Gibco-BRL, Grand Island, NY) in 20 μl of a reaction buffer that contained 50 mm Tris–HCl pH 8.3, 1 mm dithiothreitol, 75 mm KCl, 5 mm MgCl2, 25 U of RNase inhibitor (Boehringer Mannheim, Indianapolis, IN), 3.2 μg of random hexamers (Boehringer Mannheim) and 1 mm dNTPS. Five-microlitre aliquots were used for polymerase chain reaction (PCR) amplification with either human COX-1, COX-2 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific primers as previously described [19].
Double immunolabelling
Slides were blocked with 5% normal serum and incubated with 1:100 anti-COX-2 MoAb (Cayman Chemical) or polyclonal rabbit anti-COX-1 antibody (Oxford Biomedical) overnight at 4°C. Slides were developed with a standard biotin-avidin-peroxidase method (Vectastain ABC; Vector Labs, Inc., Burlingame, CA), and diaminobenzidine as substrate. Controls included slides in which primary antibody was omitted and preincubation of the primary antibody with purified COX-2 protein (Cayman Chemicals).
Identification of COX-1 or COX-2 immunoperoxidase-stained cells was performed by immunofluorescence with anti-CD68 (Dako, Copenhagen, Denmark), anti-CD3 (Ortho Diagnostic Systems) or anti-CD4 (Boehringer Mannheim). A secondary fluorescein-conjugated rabbit anti-mouse immunoglobulin was used (Dako).
The presence of activated T cells on SF smears was assessed by CD69 and CD25 immunofluorescence.
PG determination
AA-induced PG synthesis was studied in cultured Jurkat T cells, under basal or OKT3-stimulated conditions by using commercial EIA (Cayman Chemicals). Cells were treated with 30 μm AA (Sigma, St Louis, MO) for 30 min and PG determined in supernatants. We used two different antibodies, one specific for PGE2 and another with extensive cross-reactivity for most PG (recognizing PG of D, E and F series). The sensitivity of the assays was at the pm level (27 and 55 pm, respectively, for PGE).
Peripheral blood adherent mononuclear cells were cultured under the same conditions and used as positive controls for the PG EIA, non-stimulated or after 10 μg/ml LPS stimulation for 24 h.
RESULTS
Specificity of anti-COX-1 and anti-COX-2 antibodies was tested in protein extracts from adherent monocytes and purified COX-2 peptide. COX-1 but not COX-2 protein was identified in non-stimulated monocytes. LPS treatment resulted in strong COX-2 induction and suppression of COX-1 protein expression (Fig. 1a). The specificity of antibodies was further confirmed by preincubation of the antibodies with COX-2 peptide which abrogated COX-2 detection without modifying COX-1 signal in the same blots (data not shown). The presence of COX-1 and -2 proteins was analysed in monocyte-depleted PBL and cultured Jurkat cells. All showed immunodetectable COX-1 protein (Fig. 1b). COX-1 levels were higher in PBL than in cultured Jurkat T cells and did not vary after 24 h incubation with OKT3.
Fig. 1.
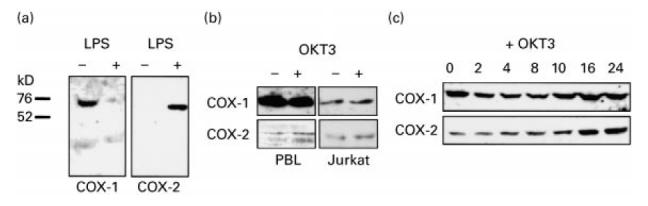
COX-1 and -2 protein expression by Western blot analysis in mononuclear cells and T cells. (a) COX-1 was expressed in non-stimulated peripheral blood monocytes and it was suppressed by 24 h of treatment with 10 μg/ml lipopolysaccharide (LPS). COX-2 was only detected in LPS-treated monocytes. (b) Non-stimulated peripheral blood T lymphocytes (PBL) expressed COX-1 at levels higher than Jurkat cells (images have been captured from the same filter). COX-2 was detected at low levels that increased by OKT3 treatment. (c) Time response of COX-2 induction with OKT3 in Jurkat cells (time points in hours).
In non-stimulated PBL, COX-2 protein was detected at very low levels that increased 3.0 ± 0.7-fold by densitometric analysis after 24 h of OKT3 stimulation (Fig. 1b). Since COX-2 protein was not detectable in adherent PBMC prior to LPS stimulation, the low level of COX-2 observed in some preparations of non-stimulated PBL was not due to monocyte contamination. This was also confirmed by FACS analysis, which excluded a significant presence of My4+ cells (< 2%). In PBL, COX-2 did not increase after LPS exposure (data not shown). The activation state of PBL was studied by CD69 FACS analysis and showed a percentage of positive cells of 17.9 ± 2.7 (non-stimulated) and 69.3 ± 13.9 (OKT3-stimulated), respectively. Jurkat cells behaved similarly and displayed a time-dependent induction of COX-2 upon OKT3 stimulation, that was maximal at 24 h reaching 4.0 ± 1.8-fold, while COX-1 levels remained unchanged (Fig. 1c).
Levels of COX-1 and COX-2 mRNA were studied in cultured Jurkat cells by reverse transcriptase (RT)-PCR analysis (Fig. 2). Both COX-1 and -2 PCR products were detected by ethidium bromide staining of PCR products on agarose gels. No changes in levels of COX-1 mRNA were observed in these cells after incubation with OKT3 antibody up to 24 h. COX-2 mRNA was detectable at very low levels prior to OKT3 stimulation and increased by 3.0 ± 0.8-fold after stimulation (Fig. 2).
Fig. 2.
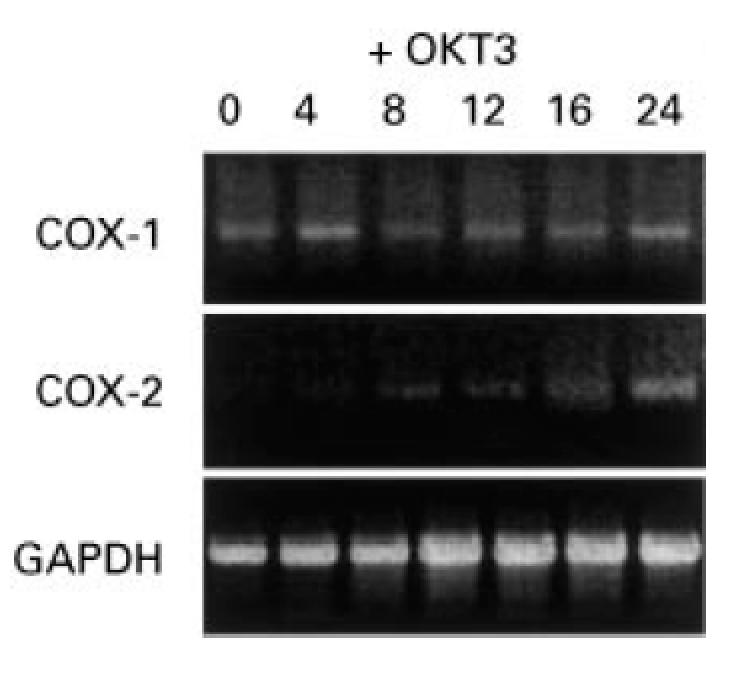
COX-1 and -2 mRNA expression in Jurkat cells by reverse transcriptase-polymerase chain reaction (RT-PCR) analysis. COX-1 mRNA was expressed in non-stimulated Jurkat cells and remained unchanged after OKT3 treatment. COX-2 was detected as a faint band in non-stimulated cells and increased after OKT3 stimulation (time points in hours). COX-1, 0.3 kb; COX-2, 0.4 kb; GAPDH, 0.6 kb.
To analyse COX-1 and -2 expression in lymphocytes in vivo, normal or RA PBMC, and RA SF cells were immunostained with COX-1 and COX-2 antibodies followed by immunofluorescent labelling with specific antibodies to CD3, CD4 or CD68 antibodies. In PBMC, COX-1 antibody produced a weak and diffuse cell staining in all cell types and COX-2 did not (data not shown). In RA SF cells, COX-1 was clearly detectable above background in many mononuclear cells that displayed two different staining intensities. Weak COX-1 staining was observed in large monocyte-macrophage CD68+ cells (Fig. 3a,c). Strong COX-1 staining was observed in a fraction of small mononuclear cells that were identified as CD3+ and CD4+ (Fig. 3a,c). COX-2 was detected by immunoperoxidase staining in few CD68+ SF mononuclear cells (Fig. 3b). No other cell types, including CD3+ and CD4+ T cells, expressed immunodetectable COX-2. The presence of abundant activated T cells in SF was confirmed by CD69 immunofluorescence (not shown), but CD25+ cells were rarely observed (< 1/500 SF cells).
Fig. 3.
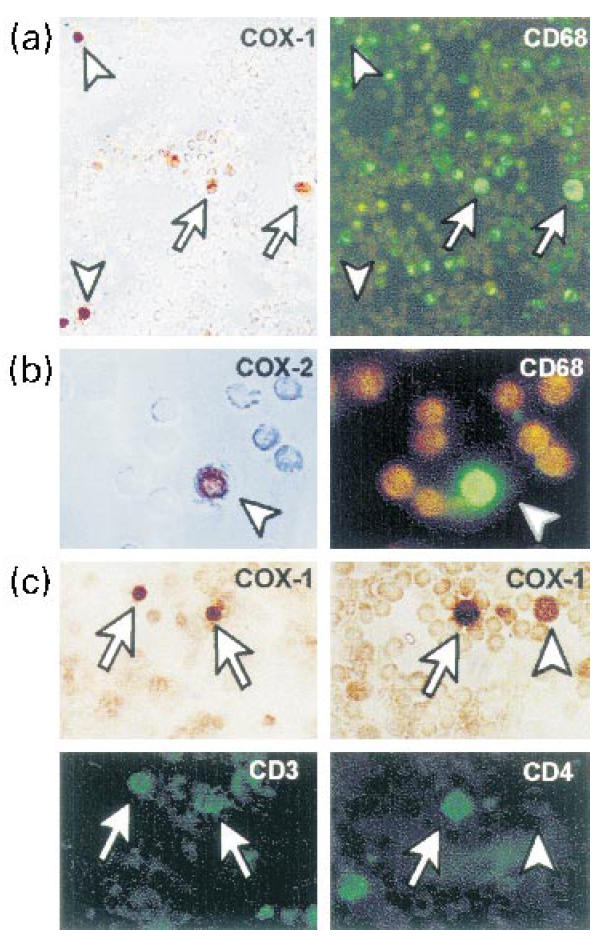
COX-1 or -2 expression by double immunoperoxidase-immunofluorescence of different cell markers in rheumatoid arthritis synovial fluid. (a) COX-1-expressing cells were identified as CD68+ monocyte-macrophage cells (arrows) or CD68− small mononuclear cells (arrowheads). (b) COX-2 was only detected in few CD68+ cells. (c) Small mononuclear cells strongly expressing COX-1 protein were identified as CD3+/CD4+ T cells.
To test the detection level of COX-2 antibody for immunohistochemistry, we compared monocytes treated in vitro with LPS with PBL treated with OKT3. In both cases, COX-2 was detectable by Western blot, although at much lower levels in PBL. LPS-treated monocytes showed a strong COX-2 immunostaining whereas OKT3-treated PBL did not stain above background (data not shown).
AA-induced PG synthesis was not detectable in undiluted supernatants of cultured Jurkat T cells, under basal or OKT3-stimulated conditions by EIA with either specific PGE2 or PG cross-reactive antibodies. In undiluted supernatants of non-stimulated and LPS-stimulated adherent mononuclear cells, PG were always detected at levels above the maximal value of the standards used (> 10 nm).
DISCUSSION
Exposure to exogenous PG has multiple inhibitory effects on T lymphocyte functions, decreasing their in vitro activation and proliferation [9–11]. In vivo, inhibition of PG synthesis with NSAID has the opposite effects and improves different states of immunodepression [20–22]. Ability of T lymphocytes to synthesize autocrine physiologically active PG has not been clearly demonstrated. Most studies do not detect COX activity in normal human T cells [14–17]. In lymphoblastoid Jurkat cells and IL-2-dependent T cell lines, some studies have demonstrated conversion of AA into minimal amounts of PGE2, probably without functional significance [12,18]. Therefore, NSAID effects on T cells were initially attributed to inhibition of exogenous PG synthesis by the monocytes present in mononuclear cell preparations [14]. Nevertheless, NSAID have also activation effects in purified lymphocytes that are independent of monocytes [12]. In our study on T cells, we did not detect physiologically significant amounts of PG after AA exposure of basal or activated cells in spite of expression of COX-1 and COX-2 mRNA and immunoreactive protein. Both COX-1 and COX-2 are regulated in T cells similarly to other cell types. COX-1 was constitutively expressed and its level remained constant after stimulation. By contrast, COX-2 was inducible upon T cell receptor triggering with OKT3 antibodies, although very low levels were detected in non-stimulated conditions. This could pertain to the presence of few activated PBL or Jurkat cells in non-;stimulated conditions. In vivo, CD4 T cells from RA SF showed immunodetectable COX-1 but not COX-2 protein. The particular status of T cell activation of RA T cells (CD69+ and CD25−) previously described [23] and confirmed in our SF samples may explain the lack of detectable COX-2. Previous studies have failed to demonstrate COX-2 in the lymphocytes infiltrating the synovium [24]. Alternatively, a low sensitivity of COX-2 antibody for immunocytochemistry may explain our findings. In support, COX-2 was detected by immunocytochemical staining of LPS-treated monocytes that exhibited levels of COX-2 much higher by Western blot analysis than stimulated T cells. In addition, in vitro OKT3-stimulated PBL were COX-2-negative by immunohistochemical staining although containing COX-2 at levels detectable by Western blot. Studies in purified T cells from different T cell-mediated diseases should demonstrate whether COX-2 is expressed in vivo during T cell activation.
The role of both COX isoforms in T cell functions remains to be defined. Lack of PG synthesis, even after COX-2 induction, is an unexpected finding. In most cell types, COX induction is followed by increase in the conversion of AA to PG. In vitro, treatment of purified T lymphocytes with COX-1 inhibitors increases cell proliferation and IL-2 production without detectable synthesis of products of 3H-labelled AA, by high performance liquid chromatography (HPLC) [12]. In addition to phospholipases and COX, PG synthases and isomerases are needed for the synthesis of the final products of this pathway. The profile of these enzymes in T cells is unknown, and therefore synthesis of other known or unknown eicosanoids cannot be excluded. An alternative explanation could be that the effects of the named COX inhibitors on T cells are due to their interaction with molecular targets different form COX. Among potential targets, disruption of signal transduction through interaction with plasma membrane proteins has been proposed [25]. In addition, COX proteins may participate on pathways other than PG synthesis through their interaction with other proteins within the endoplasmic reticulum. One example is the lymphocyte apoptosis- and autoimmunity-associated Nuc protein, whose interaction with COX can modify their cellular effects [26]. Whether NSAID can modify these interactions remains speculative.
Regardless the role of COX protein in T cells, the differential expression of both isoforms, and particularly the observed increase in COX-2 expression during T cell activation, suggests that they have different functions. Thus, the effects of selective pharmacological inhibition of COX-1 or -2 on T cell function merit further investigation.
Acknowledgments
This work was supported by a grant of the Ministerio de Educacion y Cultura (PM 96/0028). We are grateful to Juan Martin, Mercedes Bermejo and Maria Teresa Lain for expert assistance with FACS analysis and to Jose Alcami for valuable discussions.
REFERENCES
- 1.Davies P, Bailey PJ, Goldenberg MM, Ford-Hutchinson AW. The role of arachidonic acid oxygenation products in pain and inflammation. Annu Rev Immunol. 1984;2:335–7. doi: 10.1146/annurev.iy.02.040184.002003. [DOI] [PubMed] [Google Scholar]
- 2.Funk CD, Funk LB, Kennedy ME, Pong AS, Fitzgerald GA. Human platelet/erythroleukemia cell prostaglandin G/H synthase: cDNA cloning, expression, and gene chromosomal assignment. FASEB J. 1991;5:2304–12. [PubMed] [Google Scholar]
- 3.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem. 1991;266:12866–72. [PubMed] [Google Scholar]
- 4.Lee SH, Soyoola E, Chanmugam P, et al. Selective expression of mitogen-inducible cyclooxygenase in macrophages stimulated with lipopolysaccharide. J Biol Chem. 1992;267:25934–8. [PubMed] [Google Scholar]
- 5.Wilborn J, DeWitt DL, Peters-Golden M. Expression and role of cyclooxygenase isoforms in alveolar and peritoneal macrophages. Am J Physiol. 1995;268:L294–301. doi: 10.1152/ajplung.1995.268.2.L294. [DOI] [PubMed] [Google Scholar]
- 6.Geng Y, Blanco FJ, Cornelisson M, Lotz M. Regulation of cyclooxygenase-2 expression in normal human articular chondrocytes. J Immunol. 1995;155:796–801. [PubMed] [Google Scholar]
- 7.Masferrer JL, Zweiffel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclooxygenase 2 in vitro is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA. 1994;91:3228–32. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zurier RB, Quagliata F. Effect of prostaglandin E1 on adjuvant arthritis. Nature. 1971;234:304–5. doi: 10.1038/234304a0. [DOI] [PubMed] [Google Scholar]
- 9.Goodwin JS, Mesner RP, Peake GT. Prostaglandin suppression of mitogen-stimulated leukocytes in culture. J Clin Invest. 1974;54:378–83. [Google Scholar]
- 10.Rappaport RS, Dodge J. Prostaglandin E inhibits the production of human interleukin 2. J Exp Med. 1982;155:943–8. doi: 10.1084/jem.155.3.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santoli D, Phillips PD, Colt TL, Zurier RB. Suppression of interleukin 2-dependent human T cell growth in vitro by prostaglandin E (PGE) and their precursor fatty acids. Evidence for a PGE-independent mechanism of inhibition by the fatty acids. J Clin Invest. 1990;85:424–32. doi: 10.1172/JCI114455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flescher E, Fossum D, Gray PJ, Fernandes G, Harper MJK, Talal N. Aspirin-like drugs prime human T cells. Modulation of intracellular calcium concentrations. J Immunol. 1991;146:2553–9. [PubMed] [Google Scholar]
- 13.Hsia J, Sarin N, Oliver JH, Goldstein AL. Aspirin and thymosin increase interleukin-2 and interferon-gamma production by human peripheral blood lymphocytes. Immunopharmacology. 1989;17:167–73. doi: 10.1016/0162-3109(89)90045-3. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy MS, Stobo JD, Goldyne ME. In vitro synthesis of prostaglandins and related lipids by populations of human peripheral blood mononuclear cells. Prostaglandins. 1980;20:135–45. doi: 10.1016/0090-6980(80)90013-1. [DOI] [PubMed] [Google Scholar]
- 15.Goldyne ME. Lymphocytes and arachidonic acid metabolism. Prog Allergy. 1988;44:140–52. [PubMed] [Google Scholar]
- 16.Goodwin JS, Behrens T. Role of lipoxygenase metabolites of arachidonic acid in T cell activation. Ann NY Acad Sci. 1988;524:201–7. doi: 10.1111/j.1749-6632.1988.tb38543.x. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman T, Lizzio EF, Ting A, Marshall LA, Bonvini E, Jennings MK. Release of arachidonic acid metabolites by human monocytes or lymphocytes: effect of treatment with interferon on stimulation by phorbol ester or calcium ionophore. Clin Immunol Immunopathol. 1987;44:82–92. doi: 10.1016/0090-1229(87)90054-7. [DOI] [PubMed] [Google Scholar]
- 18.Aussel C, Mary D, Fehlmann M. Prostaglandin synthesis in human T cells: its partial inhibition by lectins and anti-CD3 antibodies as a possible step in T cell activation. J Immunol. 1987;138:3094–9. [PubMed] [Google Scholar]
- 19.In˜iguez MA, Pablos JL, Carreira PE, Cabre F, Gómez-Reino JJ. Detection of COX-1 and COX-2 isoforms in synovial fluid cells from inflammatory joint diseases. Br J Rheumatol. 1998;37:773–8. doi: 10.1093/rheumatology/37.7.773. [DOI] [PubMed] [Google Scholar]
- 20.Ambrus JL, Jr, Haneiwich S, Chesky L, McFarland P, Engler RJ. Improved in vitro antigen-specific antibody synthesis in two patients with common variable immunodeficiency taking an oral cyclooxygenase and lipoxygenase inhibitor (ketoprofen) J Allergy Clin Immunol. 1991;88:775–83. doi: 10.1016/0091-6749(91)90185-q. [DOI] [PubMed] [Google Scholar]
- 21.Blomgren H, Rotstein S, Wasserman J, Petrini B, Hammarstrom S. In vitro capacity of various cyclooxygenase inhibitors to revert immune suppression caused by radiation therapy for breast cancer. Radiother Oncol. 1990;19:329–35. doi: 10.1016/0167-8140(90)90033-s. [DOI] [PubMed] [Google Scholar]
- 22.Wachsman M, Aurelian L, Burnett JW. The prophylactic use of cyclooxygenase inhibitors in recurrent herpes simplex infections. Br J Dermatol. 1990;123:375–80. doi: 10.1111/j.1365-2133.1990.tb06298.x. [DOI] [PubMed] [Google Scholar]
- 23.Hernandez-Garcia C, Fernandez-Gutierrez B, Morado I, Ban˜ares AA, Jover JA. The CD69 activation pathway in rheumatoid arthritis synovial fluid T cells. Arthritis Rheum. 1996;39:1277–86. doi: 10.1002/art.1780390803. [DOI] [PubMed] [Google Scholar]
- 24.Siegle I, Klein T, Backman JT, Saal JG, Nusing RM, Fritz P. Expression of cyclooxygenase 1 and cyclooxygenase 2 in human synovial tissue: differential elevation of cyclooxygenase 2 in inflammatory joint diseases. Arthritis Rheum. 1998;41:122–9. doi: 10.1002/1529-0131(199801)41:1<122::AID-ART15>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 25.Abramson S, Korchak H, Ludewig R, et al. Modes of action of aspirin-like drugs. Proc Natl Acad Sci USA. 1985;82:7227–31. doi: 10.1073/pnas.82.21.7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ballif BA, Mincek NV, Barratt JT, Wilson ML, Simmons DL. Interaction of cyclooxygenases with an apoptosis- and autoimmunity-associated protein. Proc Natl Acad Sci USA. 1996;93:5544–9. doi: 10.1073/pnas.93.11.5544. [DOI] [PMC free article] [PubMed] [Google Scholar]