

Abstract
Vascular endothelial growth factor (VEGF) is abundant in synovium and synovial fluids, where it probably contributes to vascular permeability and angiogenesis in arthritic joints. To investigate the probable sources of VEGF in synovium, we compared the ability of several cytokines (TGF-β, platelet-derived growth factor (PDGF), IL-1, tumour necrosis factor (TNF), basic fibroblast growth factor (bFGF) that are associated with arthritis and angiogenesis, to stimulate secretion of VEGF protein by human synovial fibroblasts. TGF-β was the strongest inducer of VEGF secretion; six times more VEGF was secreted when cells were stimulated by TGF-β than when stimulated by PDGF or IL-1 for 24 h. TNF-α and bFGF did not stimulate any secretion of VEGF. The stimulatory effects of TGF-β and IL-1 on VEGF secretion were additive. Hypoxic culture alone also stimulated VEGF secretion, but more importantly, hypoxic culture conditions doubled the rate of VEGF secretion stimulated by the cytokines TGF-β and IL-1. When dermal and synovial fibroblasts were stimulated identically by hypoxia and cytokines (TGF-β and IL-1), synovial fibroblasts secreted four times more VEGF than did dermal fibroblasts. Thus in rheumatoid arthritis, the capacity of synovial fibroblasts in the hypoxic environment to secrete large amounts of VEGF in response to cytokines such as TGF-β probably contributes significantly to angiogenesis in the synovium.
Keywords: arthritis, angiogenesis, cytokines, transforming growth factor-beta, hypoxia, VEGF
INTRODUCTION
Angiogenesis is thought to be an essential process in the development of arthritis [1–3]. Angiogenesis supports the extensive vascularization that occurs during development of the synovial pannus and synovial villi in chronic rheumatoid arthritis (RA). Antagonism of angiogenesis may provide a therapeutic strategy in the treatment of RA. It is therefore of interest to define the angiogenic factors that contribute to arthritic pathology and to understand the regulation of their production.
Recent studies have established the importance of a substance termed vascular endothelial growth factor (VEGF) to both developmental vasculogenesis and to angiogenesis that occurs in normal and pathologic tissue development [4,5]. We and others have shown that VEGF is present in synovial fluids in significant amounts, and it is expressed in inflamed synovium, where it is thought to be produced by synovial fibroblasts and activated macrophages in RA [2,3,6,7]. VEGF is a selective endothelial mitogen and vascular permeability factor, that is generally inducible by hypoxia and is important in both normal and pathologic processes [5], such as embryonic development [8], wound healing [9], and solid and ascites tumour formation [10,11].
Because of the abundance of cytokines in the chronically inflamed synovium in RA [12,13] and because hypoxic conditions are thought to exist in arthritic joints [14,15], we examined whether selected cytokines and hypoxic culture directly induced VEGF protein secretion by primary cultures of human synovial cells. We report here that TGF-β is an exceptionally strong stimulator of VEGF production by synovial fibroblasts, compared with platelet-derived growth factor (PDGF), IL-1 and hypoxia, and that cells secrete even greater amounts of VEGF when exposed simultaneously to hypoxic conditions and cytokines.
MATERIALS AND METHODS
Cytokines (human recombinant TGF-β1, tumour necrosis factor-alpha (TNF-α), IL-1α, basic fibroblast growth factor (bFGF), PDGF) were purchased from R&D Systems, Inc. (Rochester, MN). The ELISA kit for VEGF was purchased from R&D Systems. Kits for random-primed labelling reactions were purchased from Promega (Madison, WI).
Primary cell lines
Synovial fibroblasts were cultured from human rheumatoid synovial tissue removed during surgical procedures. Tissue was minced, digested overnight with collagenase, separated from undigested tissue by unit gravity sedimentation and low-speed centrifugation. The suspended cells were pelleted, washed several times in RPMI medium containing 10% fetal calf serum (FCS) and plated at high density into T-25 culture flasks. After overnight incubation, non-adherent cells were removed by changing the medium, and the attached cells were cultured in RPMI with 10% FCS through three to five addition passages over several weeks before they were used in experiments. The cultures became increasingly enriched in proliferating fibroblastic cells and by week 5 homogeneous cultures of fibroblastic morphology were obtained. Cultures of synovial fibroblasts became confluent at comparatively low cell numbers (1–2 × 106 cells/T-75) relative to primary dermal fibroblasts (5 × 106/T-75), and appeared slightly refractile at the edges.
Dermal fibroblasts were cultured from human foreskin tissue digested with collagenase and plated in RPMI 1640 10% FCS medium and used after several weeks of culture. Dermal fibroblasts were morphologically distinct from synovial fibroblasts, grew more rapidly and reached higher cell density at confluence than did synovial fibroblasts.
ELISA measurement of secreted VEGF
Confluent monolayers of synovial fibroblasts were preincubated for 48 h with fresh serum-free medium before each experiment, unless otherwise noted in the figure legend. Serum contains factors that induce VEGF secretion (TGF-β, PDGF), and preliminary experiments showed that basal VEGF production continued for approx. 48 h after withdrawal of serum containing medium. To start an experiment, the cells were changed to fresh medium at time zero and cytokines were added. After incubation for varying lengths of time, either with or without exogenous cytokines or hypoxic culture, the medium was removed and was stored at −80°C until assayed. In preliminary experiments, an antibody-based fluorescence assay was used to measure VEGF [16]. For all the data presented here, VEGF concentrations were determined using a commercial ELISA kit, exactly according to instructions (R&D Systems). Triplicate samples were analysed for all conditions examined and the means and standard errors calculated and plotted.
Hypoxic cell culture
Hypoxic culture was performed by incubating cells in an incubator (Napco, Chicago, IL) held at 3% oxygen, by the addition of nitrogen. Serum-free medium was added to confluent T-75 flasks for 48 h prior to hypoxic culture, and then fresh medium (5 ml) was added to each confluent T-75 flask immediately before placing into hypoxic culture. After 6, 24 or 48 h, the medium was collected and total RNA isolated from the cells. Cell viability was determined by trypan blue exclusion and by continuing culture of the cells after hypoxia for up to 72 h.
Isolation of RNA
Medium was removed from cells and total mRNA was isolated using the RNAzol method (Ambion, Friendswood, TX) or Totally RNA (Ambion, Austin, TX), exactly as directed. Yields from synovial fibroblasts ranged from 10 to 20 μg/T-75 flask of confluent cells. The quality and quantity of the RNA was assessed by electrophoresis on agarose gels and by optical density (OD) at 260 nm.
RNase protection assay
An assay was developed to distinguish the transcripts that code for each of the three major VEGF isoforms (VEGF121, VEGF165, VEGF189). A cDNA probe coding for 377 base pairs of the human VEGF189 sequence was cloned between the EcoRI and BamHI sites of plasmid pGem3Zf(+) Vector (Promega) [17]. Single-stranded 32P-labelled cRNA probes were prepared by in vitro transcription (Ambion) and the 420-base labelled material purified by electrophoresis through polyacrylamide/urea gels and isolation from the gel. An actin-specific cRNA probe (Ambion) was used to assess the amounts of total RNA in samples, in a parallel assay. As controls, two 10-μg aliquots of yeast RNA were hybridized with the same amount of probe, with and without addition of RNase. After overnight hybridization of 10 μg total RNA for VEGF analysis or 2 μg total RNA for actin transcript analysis, the non-hybridized labelled probe was digested with a mixture of RNase A and RNase T, and the resulting 32P-labelled products of different lengths were separated on 6% polyacrylamide/8M urea gels. Radioactive bands thus arose from protection of the 420-base labelled probe from digestion by RNase, by hybridization with VEGF121,VEGF165 or VEGF189 transcripts present in the total RNA. Hybridization with VEGF121 resulted in protected bands of 144 and 29 base pairs, hybridization with VEGF165 resulted in bands of 276 and 29 base pairs and with VEGF189 resulted in bands of 377, 347 and 30 base pairs. Note that the predicted 377 band resulting from protection by the VEGF189 transcript actually appears as a doublet; the VEGF189 transcript contains a AU-rich sequence near the end that is sensitive to RNase digestion, and due to weak hybridization there results a partial digestion by RNases, to yield the slightly shorter form. Visualization of protected radioactive bands was by x-ray film and the relative intensity of radioactive bands was quantified either from the films with a Microscan 1000 2D Densitometer (TRI Industries, Nashville, TN) or directly from the dried gels with a Phosphorimager (Molecular Dynamics, Sunnyvale, CA). Transcripts of known size were generated by in vitro transcription from RNA Century Marker Template (Ambion) and were used as size markers.
Northern blotting
A 520-bp VEGF-specific hybridization probe was prepared by performing reverse transcriptase-polymerase chain reaction (RT-PCR) using total mRNA from TGF-β-stimulated human synovial fibroblasts and a VEGF primer pair [18]. RT-PCR with this primer pair yields cDNA for several isoforms of VEGF. The 520-bp amplified DNA product was isolated from a 2% agarose gel. The isolated cDNA was sequenced to confirm that it matched the published VEGF sequence. This 520-bp cDNA corresponds to the VEGF121 isoform, but will recognize all VEGF transcripts. 32P-labelled hybridization probe was prepared using 25 ng of the 520-bp DNA and random primer labelling (Prime-A-Gene Kit; Promega). Northern blotting was performed as described using 10 μg of total RNA [17]. Briefly, Northern blots were hybridized overnight at 65°C with 2 × 106 ct/min per ml 32P-labelled DNA and the blots were washed twice in 1× SSC/5% SDS at 50°C and once at 55°C. To quantify RNA loading, the blots were hybridized with a 32P-labelled cDNA designed to hybridize with 28S ribosomal RNA (pTri-RNA-28S,; Ambion).
RESULTS
Comparison of the induction of VEGF by different cytokines
To compare the relative capacity of cytokines to induce VEGF secretion by human synovial fibroblasts, selected cytokines (bFGF, TNF, PDGF, IL-1, TGF-β) were added to serum-free culture medium at 10 ng/ml. After different incubation times, aliquots of medium were removed and assayed by ELISA for VEGF. Serum-free culture conditions were required since serum contains VEGF-stimulatory substances; 48 h of serum starvation was necessary to reduce VEGF mRNA and VEGF protein secretion to their lowest levels (data not shown). The total amount of VEGF secreted into medium during 2, 6, 12 and 24 h was determined by ELISA; note that cell-associated VEGF isoforms (i.e. VEGF189) would not be determined in this assay. As shown in Fig. 1, some of the cytokines tested stimulated secretion of significantly more VEGF protein than was secreted by unstimulated synovial fibroblasts (TGF-β, IL-1 and PDGF) and the concentration of VEGF in medium increased steadily with time. The data in Fig. 1 are taken from a single experiment to allow a direct quantitative comparison of the responses to individual cytokines with a single primary cell culture, but the data shown are representative of the results observed in at least six experiments performed with synovial cell cultures obtained from four different donors and examined periodically over a period of 1 year.
Fig. 1.
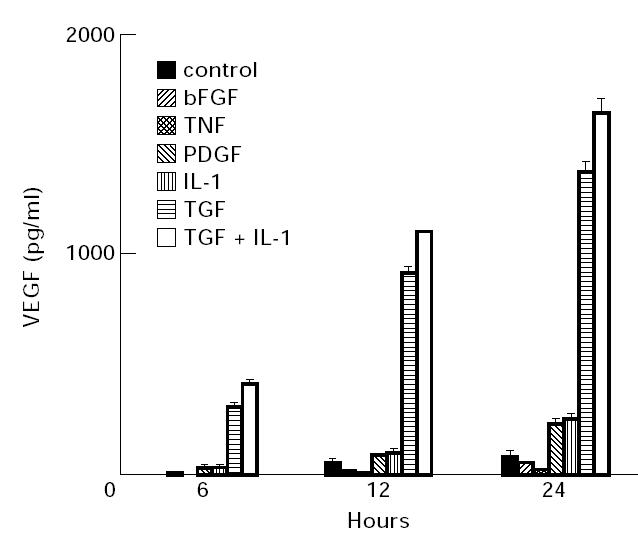
Secretion of vascular endothelial growth factor (VEGF) by synovial fibroblasts stimulated with cyokines. The cytokines indicated were added to replicate wells containing confluent monolayers of human synovial fibroblasts (approx. 20 000 cells/well in 12-well plates) in 0.5 ml of serum-free medium. All cytokines were added at 10 ng/ml into triplicate wells to ensure saturating concentrations. After 2, 6, 24 and 48 h of culture, medium was removed and the concentration of VEGF attained in each well determined by ELISA analysis. In all cases VEGF was undetectable in medium after 2 h of culture (data not shown). The mean of triplicate wells and the standard error are shown. These data are representative of six experiments performed with cell lines derived from tissue from four different patients. TNF, Tumour necrosis factor; PDGF, platelet-derived growth factor.
TGF-β was clearly the strongest inducer of VEGF secretion, while IL-1 and PDGF also induced a level of secretion higher than controls. The strong induction of VEGF secretion by TGF-β was a consistent response, and was independent of cell passage number. The combination of TGF-β and IL-1 generally stimulated an additive response, as shown in Fig. 1. However, in one experiment with early passage cells, the combination of TGF-β and IL-1 produced a synergistic induction of VEGF secretion during 24 h (data not shown). Compared with stimulation by TGF-β, PDGF induced a much lower level of VEGF secretion, as did IL-1. TGF-β consistently stimulated secretion of much more VEGF than the other cytokines, ranging from four to six times the amount of VEGF induced by IL-1 or PDGF, despite similar kinetics of induction of VEGF mRNA for TGF-β and IL-1 (see Figs 2 and 3). Some cytokines tested did not stimulate any VEGF secretion, as shown in Fig. 1 for bFGF and TNF.
Fig. 2.
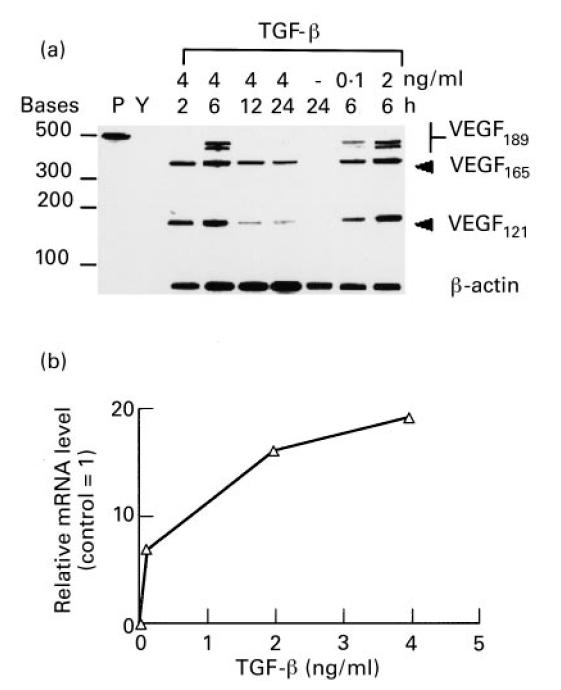
Kinetics and dose–response of induction of mRNA for vascular endothelial growth factor (VEGF) isoforms by TGF-β. Confluent cultures of synovial fibroblasts were incubated for 48 h in serum-free medium. TGF-β was then added to fresh medium at various concentrations for the times indicated. Total RNA was isolated and 5 μg per sample were analysed by an RNase protection assay designed to detect individual VEGF isoforms. Two micrograms of total RNA were used for actin transcript protection to assess whether equal RNA was present in each sample. (a) Autoradiograph of a polyacrylamide gel. Lane P, radioactive probe alone; lane Y, control with 5 μg yeast RNA. TGF-β was added to cell cultures at 4 ng/ml for 2, 6, 12 and 24 h, at 0.1, 2 and 4 ng/ml for 6 h, or was not added as indicated above. VEGF isoforms of 189, 165 and 121 amino acids are indicated. (b) Densitometric analysis of VEGF165 bands induced by TGF-β after 6 h and expressed relative to VEGF165 transcripts of cells incubated 24 h without TGF-β.
Fig. 3.
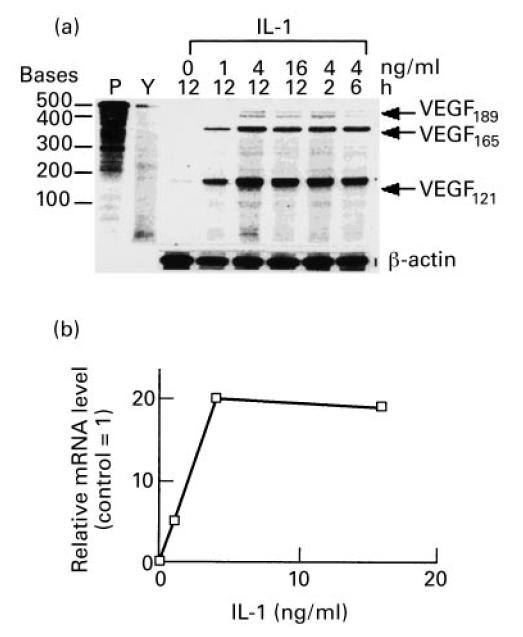
Kinetics and dose–response of induction of mRNA for vascular endothelial growth factor (VEGF) isoforms by IL-1. (a) Confluent cultures of synovial fibroblasts were incubated for 48 h in serum-free medium. IL-1 was then added to fresh medium at various concentrations for the times indicated. Total RNA was isolated and 5 μg per sample were analysed by an RNase protection assay designed to detect individual VEGF isoforms. Two micrograms of total RNA was used for actin transcript protection to assess whether equal RNA was present in each sample. Autoradiograph of a polyacrylamide gel. Lane P, radioactive probe alone; lane Y, control with 5 μg yeast RNA. IL-1 was added to cell cultures at 0, 1, 4 and 16 ng/ml for 12 h, or 4 ng/ml for 2, 6 and 12 h as indicated above. VEGF isoforms of 189, 165 and 121 amino acids are indicated. (b) Densitometric analysis of VEGF165 bands induced by various doses of IL-1 after 12 h and expressed relative to VEGF165 transcripts of cells incubated for 24 h without IL-1.
Induction of individual VEGF isoforms by cytokines
To determine whether there was selectivity in the VEGF isoforms induced by the cytokines TGF-β and IL-1, a quantitative RNase protection assay was performed on total RNA isolated from cytokine-stimulated synovial fibroblasts. The RNase protection assay was designed to generate three major radioactive bands of differing lengths when protected from RNase digestion by hybridization with transcripts for each of the three major VEGF isoforms, VEGF165, VEGF121 and VEGF189 [17].
As shown in Fig. 2 for TGF-β and Fig. 3 for IL-1, there was no selective induction observed for any particular isoform of VEGF. Stimulation with either cytokine resulted in an essentially equal induction of the two major secreted isoforms of VEGF (VEGF165, VEGF121) and the cell-associated isoform (VEGF189). Induction of the VEGF189 isoform by TGF-β appeared to be more transient than the other isoforms, its expression peaking at 6 h (see Fig. 2). The relative induction of VEGF121 appeared greater for IL-1 than for TGF-β (see Fig. 3), although despite this fact, the total amount of secreted VEGF protein detected by ELISA was much lower after 24 h of stimulation with IL-1 than with TGF-β (see Fig. 1).
Kinetics and dose–response of VEGF mRNA induction by TGF-β and IL-1
The RNase protection assay was also used to determine the dose–responses of synovial fibroblasts to varying amounts of TGF-β and IL-1. As seen in Fig. 2a,b, doses of TGF-β ranging from 0.1 to 4 ng/ml increased VEGF mRNA well above unstimulated controls. As shown in Fig. 2b, densitometric analysis of the bands for the VEGF165 isoform indicated a maximal induction of approx. 20-fold compared with unstimulated cells, which occurred with 4 ng/ml of TGF-β in 6 h. At this saturating dose of TGF-β, the levels of VEGF mRNA were induced above control levels within 2 h of addition to the cultures, with a maximum induction seen after 6 h of incubation. For IL-1, a dose–response was also seen, with no further increase in levels of VEGF mRNA observed at concentrations > 4 ng/ml, shown in Fig. 3a,b. The increase of VEGF mRNA levels of VEGF165 specifically was quantified by densitometry. IL-1 induced increases in VEGF mRNA from 2 to 12 h of incubation, and with IL-1 at 4 ng/ml, VEGF mRNA was induced approx. 20-fold.
VEGF mRNA quantified by Northern hybridization
Since TGF-β and IL-1 are probably present simultaneously in arthritic synovium, we examined each alone and both together for the ability to induce steady-state VEGF mRNA levels. Both TGF-β and IL-1 when added alone at saturating concentrations induced VEGF mRNA steady-state levels as shown by Northern blotting in Fig. 4. In addition, there was a dramatic synergistic response in the induction of VEGF mRNA when TGF-β and IL-1 were added together for 6 h in cultures of synovial fibroblasts. Quantitative densitometry indicated an induction of approximately 25-fold in the level of VEGF mRNA when both cytokines were present, compared with inductions of approximately four-fold for either cytokine alone.
Fig. 4.
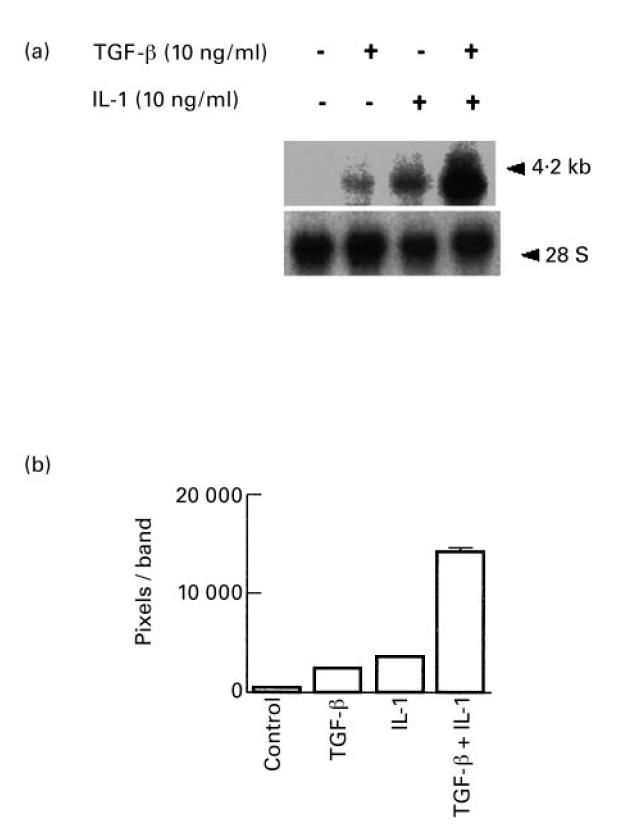
Induction of vascular endothelial growth factor (VEGF) mRNA expression by TGF-β and IL-1. Synovial fibroblasts were grown to confluence in T-75 flasks, incubated for another 48 h in serum-free medium and then fresh medium was added with or without TGF-β, IL-1 or both combined (10 ng/ml). After 6 h at 37°C total RNA was isolated. (a) Northern hybridization. Total RNA (10 μg/lane) and RNA size standards were electrophoresed and blotted onto nylon membranes and hybridized with a VEGF-specific 32P-labelled cDNA. The VEGF transcript is seen at 4.2 kb. The blot was stripped and rehybridized with a 28S RNA-specific labelled probe (28S). (b) Densitometric analysis. The bands on exposed films were imaged and digitized and the number of pixels associated with each band quantified. Induction of VEGF mRNA transcripts in cells treated with TGF-β and IL-1 combined for 6 h was increased approx. 25-fold above control cells.
While the induction of VEGF mRNA by TGF-β and IL-1 together appeared synergistic, an ELISA analysis of the amount of VEGF secreted by cells indicated that when combined, TGF-β and IL-1 stimulated an additive increase (see Fig. 1). Occasionally, the combination of TGF and IL-1 produced a synergism in the amount of VEGF secreted; for example, in one experiment TGF-β alone induced 50 pm secreted VEGF in 24 h, and IL-1 stimulated 25 pm VEGF in 24 h, but together TGF-β + IL-1 stimulated 150 pm secreted VEGF. The magnitude of the overall response to the combination of TGF and IL-1 varied somewhat with cells derived from different patients as well as the age of the cells in culture. Results very similar to those in Fig. 1 (data not shown) were obtained with four different synovial fibroblast preparations derived from different donors (20–70 years of age). Note also that the cell-associated isoform VEGF189 is not measured by ELISA of culture medium, but is probably induced by TGF-β and IL-1, as shown in Figs 2 and 3, and therefore VEGF189 transcripts would contribute to the total VEGF mRNA detected in the Northern blot analysis shown in Fig. 4, but not to VEGF protein measured by ELISA.
Hypoxia-induced VEGF mRNA and protein secretion
VEGF mRNA levels and secretion by synovial fibroblasts were compared after the cells were incubated under hypoxic (3% oxygen) versus normoxic (21% oxygen) conditions for 48 h. In hypoxic cells VEGF mRNA levels increased eight-fold above control levels within 48 h and three-fold more VEGF protein was secreted by hypoxic cells than by normoxic cells in 48 h (1500 pg/ml versus 500 pg/ml, respectively; 106 cells/flask with 3.0 ml serum-free medium) (data not shown).
Hypoxia augments cytokine-induced VEGF secretion
In vivo, synovial cells are probably exposed simultaneously to cytokines and hypoxic conditions. Therefore, by Northern blotting we determined the effect of hypoxia on VEGF mRNA induction by the cytokines TGF-β and IL-1. As shown in Fig. 5, after 6 h of incubation, when cytokine induction of VEGF mRNA was very strong, hypoxia induced no further increase of VEGF mRNA levels. However, after 24 h of incubation, when the induction of VEGF mRNA by cytokines had waned, hypoxia greatly enhanced VEGF mRNA levels compared with the level induced by the cytokines in normoxic cultures. Without cytokines present, hypoxia induced only moderate increases m VEGF mRNA, both at 6 h and at 24 h, also shown in Fig. 5.
Fig. 5.
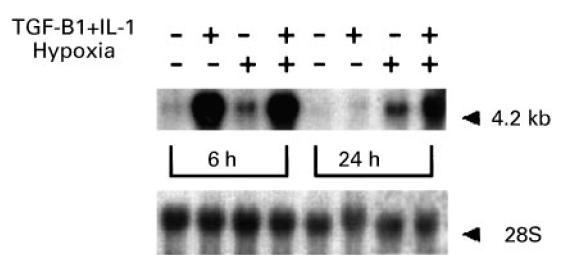
Combined effect of hypoxic culture and cytokines on vascular endothelial growth factor (VEGF) mRNA levels. After 48 h of culture in serum-free medium, confluent fibroblast cultures were changed to fresh medium with or without addition of TGF-β (10 ng/ml) and IL-1 (10 ng/ml) added together. Replicate flasks were cultured under either normoxic (21% oxygen) or hypoxic (3% oxygen) conditions for 6 h or 24 h. Total RNA was isolated from cells in six flasks in each case, and Northern hybridization for VEGF performed with 5 μg total RNA. The blot was rehybridized for 28S RNA to assess equality of loading The absence or presence of cytokines, the incubation time period and normoxic or hypoxic culture are indicated in the figure.
The augmentation of VEGF mRNA by hypoxia and cytokines was accompanied by a parallel increase in VEGF protein secretion, as shown in Fig. 6, for both synovial and dermal fibroblasts. Hypoxia enhanced the amount of VEGF released by cytokine-stimulated synovial fibroblasts, doubling the amount of VEGF secreted by the cells during 12–72 h of incubation. Culture of synovial fibroblasts with either cytokines alone or hypoxic conditions alone also increased VEGF secretion to approximately the same extent above control levels. In contrast, we could not detect any VEGF secreted by unstimulated dermal fibroblasts or when dermal fibroblasts were cultured under hypoxic conditions alone, but the addition of cytokines stimulated VEGF secretion and hypoxia further enhanced the cytokine-stimulated VEGF production, as shown in Fig. 6.
Fig. 6.
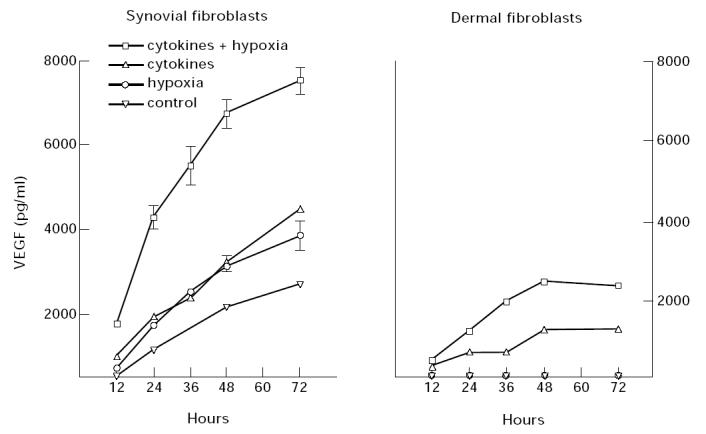
Effect of hypoxia on cytokine-induced vascular endothelial growth factor (VEGF) secretion. Synovial fibroblasts and dermal fibroblasts were grown to confluence in 48-well plates, cultured in the absence of serum for 24 h and cytokines (TGF-β and IL-1) were added at 10 ng/ml to fresh serum-free medium. Cells were then incubated in either normoxic (21% oxygen) or hypoxic conditions (3% oxygen). At the times indicated, the medium was collected from triplicate wells and assayed by ELISA for secreted VEGF. The data were normalized to constant cell number (5 × 103) and the mean concentration of VEGF and the s.e.m. are shown.
Interestingly, when compared on a ‘per cell’ basis, synovial fibroblasts made about four times more VEGF than did dermal fibroblasts, when stimulated in an identical manner, as shown in Fig. 6. Synovial fibroblasts simultaneously stimulated with cytokines and hypoxia released enough VEGF in 72 h to reach approximately 8 ng/ml, versus only 2 ng/ml secreted by dermal fibroblasts. We do not yet understand why synovial fibroblasts have a greater capacity to secrete VEGF when compared with dermal fibroblasts.
DISCUSSION
The presence of VEGF in arthritic synovium strongly suggests its participation in synovial pathogenesis [6,7], probably by promoting angiogenesis, vascular permeability [4] and microvessel stability in synovium [19]. Since VEGF appears to be a very important angiogenesis factor in vivo, the factors that regulate VEGF production by different cell types are of great interest. TGF-β has previously been shown to induce VEGF mRNA expression and protein secretion in established cell lines [20], but here we show that primary cultures of synovial fibroblasts are very strongly induced by TGF-β to release VEGF. In an early report, IL-1 and prostaglandin were shown to induce VEGF mRNA in synovial fibroblasts, but the synthesis and secretion of VEGF protein were not demonstrated [21]. A more recent study showed that IL-1 induces VEGF protein secretion by synovial cells [22], but other cytokines were not studied or compared. Here we directly compared the responses of primary synovial fibroblasts to different cytokines, and we found that TGF-β was an extremely strong stimulator of VEGF production by synovial fibroblasts, and that TGF-β could also combine with IL-1 stimulation and hypoxia to stimulate very large amounts of VEGF secretion. The high levels of TGF-β (10–40 ng/ml) found in arthritic synovial fluids and tissue [23] suggest that TGF-β is important in the regulation of VEGF production in arthritic synovial tissue [3,6,7].
Inflamed arthritic synovium is thought to experience hypoxic conditions [3], resulting from several factors, including the pressure exerted on capillaries that results from tissue oedema (obstructing vascular flow in the synovium), a dearth of superficial capillaries in arthritic synovium compared with normal synovium [24], and damage to tissue caused by reactive oxygen species [14]. Another indication of hypoxia in arthritic joints is that lactate, a product of glycolytic metabolism (anaerobic metabolism), accumulates faster in lavage fluid from arthritic joints compared with normal joints [15]. For these reasons we examined the effect of hypoxia on VEGF production by synovial cells. We have confirmed that hypoxia stimulates VEGF secretion by synovial fibroblasts [22], but more importantly, we showed that hypoxia substantially augments cytokine stimulation of VEGF production. Thus it is the combined effect of hypoxia and cytokines on synovial fibroblasts in arthritic joint tissue which may be especially important in driving synovial angiogenesis via local VEGF production.
In addition to synovial fibroblasts, it is likely that some VEGF in synovium derives from monocytes and activated macrophages, both of which have been shown to produce VEGF [25,26]. The factors which regulate the production of VEGF by macrophages and monocytes remain to be elucidated. In results not presented, we found that human monocytes, cultured for 20 days to induce macrophage surface markers, did not secrete enough VEGF to detect by ELISA, even after the medium was concentrated 20-fold (data not shown). However, immunostaining in human synovium suggests that VEGF is indeed produced by macrophage-like cells in human arthritic synovium [6]. Thus it remains unclear what factors play a role in inducing VEGF production by macrophages in vivo.
Our study and others [21] clearly indicate that the synovial fibroblast is a potential source of VEGF in arthritic synovium. Moreover, based on a comparison with primary dermal fibroblasts, fibroblasts originating from inflamed synovium have a significantly greater capacity to secrete VEGF in response to inflammatory cytokines and hypoxia.
VEGF thus appears to be an important angiogenic factor likely to contribute to synovial hyperplasia by stimulating growth of the vascular system required for the outgrowth of synovial villi which occurs in chronic arthritis. In addition to playing a role in oxygenating and supporting the nutritional requirements of this new synovial tissue, such new vessels offer additional endothelial surface area for capture of inflammatory and immune cells. Thus VEGF, VEGF receptors and VEGF-regulatory factors in synovium all represent useful therapeutic targets that could potentially reduce pannus formation, inflammatory cell infiltration and the development of the lymphoid character of synovium in chronic rheumatoid arthritis.
Acknowledgments
This study was supported by the Arthritis Foundation, V.A. Merit Review and Hitchcock Foundation Grants to R.A.F.
REFERENCES
- 1.Peacock DJ, Banquerigo ML, Brahn E. Angiogenesis inhibition suppresses collagen arthritis. J Exp Med. 1992;175:1135–8. doi: 10.1084/jem.175.4.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koch A. Angiogenesis: implications for rheumatoid arthritis. Arthritis Rheum. 1998;41:951–62. doi: 10.1002/1529-0131(199806)41:6<951::AID-ART2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 3.Paleolog E, Fava RA. Angiogenesis in rheumatoid arthritis. Spring Sem Immunol. 1998 doi: 10.1007/BF00832000. in press. [DOI] [PubMed] [Google Scholar]
- 4.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hypermeability and angiogenesis. Am J Pathol. 1995;146:1029–39. [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrara N, Davis-Srnyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 6.Fava RA, Olsen NJ, Spencer-Green G, et al. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med. 1994;180:341–6. doi: 10.1084/jem.180.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch AE, Harlow LA, Haines GK, Amento EP, Unemori EN, Lee Wong W, Pope RM. Vascular endothelial growth factor: a cytokine modulating endothelial function in rheumatoid arthritis. J Immunol. 1994;152:4149–56. [PubMed] [Google Scholar]
- 8.Ferrara N, Carver-Moore K, Chen H, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 9.Detmar M, Yeo K-T, Nagy JA, et al. Keratinocyte-derived vascular permeability factor (vascular endothelial growth factor) is a potent mitogen for dermal microvascular endothelial cells. J Invest Dermatol. 1995;105:44–50. doi: 10.1111/1523-1747.ep12312542. [DOI] [PubMed] [Google Scholar]
- 10.Millauer B, Shawer LK, Plate KH, Risau W, Ullrich A. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367:576–9. doi: 10.1038/367576a0. [DOI] [PubMed] [Google Scholar]
- 11.Senger DR, Galli SJ, Dvorak AM, Peruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–5. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 12.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Ann Rev Immunol. 1996;21:397–439. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 13.Firestein GS, Zvafiler NJ. Inflammation. 2. New York: Raven Press; 1992. [Google Scholar]
- 14.Allen RE, Blake DR, Nazhat NB, Jones P. Superoxide radical generation by inflamed human synovium after hypoxia. Lancet. 1989;8657:282–3. doi: 10.1016/s0140-6736(89)90476-5. [DOI] [PubMed] [Google Scholar]
- 15.James MJ, Cleland LG, Rofe AM. Determinants of synovial fluid lactate concentration. J Rheum. 1992;19:1107–10. [PubMed] [Google Scholar]
- 16.Yeo KT, Sioussat TM, Faix JD, Senger DR, Yeo TK. Development of time-resolved immunofluorometric assay of vascular permeability factor. Clin Clern. 1992;38:71–75. [PubMed] [Google Scholar]
- 17.Detmar M, Brown LF, Berse B, Jackman RW, Elicker BM, Dvorak BF, Claffey K. Hypoxia regulates the expression of vascular permeability factor vascular endothelial growth factor (VPF/VEGF) and its receptors in human skin. J Invest Dermatol. 1997;108:263–8. doi: 10.1111/1523-1747.ep12286453. [DOI] [PubMed] [Google Scholar]
- 18.Ballaun C, Weninger W, Uthman A, Weich H, Tschachler E. Human keratinocytes express the three major splice forms of vascular endothelial growth factor. J Invest Dermatol. 1995;104:7–10. doi: 10.1111/1523-1747.ep12613450. [DOI] [PubMed] [Google Scholar]
- 19.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor vascular permeability factor antibody. Proc Natl Acad Sci USA. 1996;93:14765–70. doi: 10.1073/pnas.93.25.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pertovaara L, Kaipainen A, Mustonen T, Orpana A, Ferrara N, Saksela O, Alitalo K. Vascular endothelial growth factor is induced in response to transforming growth factor-β in fibroblastic and epithelial cells. J Biol Chem. 1994;269:6271–4. [PubMed] [Google Scholar]
- 21.Ben-Av P, Crofford LJ, Wilder RL, Hla T. Induction of vascular endothelial growth factor expression in synovial fibroblasts by prostaglandin E and interleukin-1: a potential mechanism for inflammatory angiogenesis. FEBS Letters. 1995;372:83–7. doi: 10.1016/0014-5793(95)00956-a. [DOI] [PubMed] [Google Scholar]
- 22.Jackson JR, Minton JAL, Ho ML, Wei N, Winkler JD. Expression of vascular endothelial growth factor in synovial fibroblasts is induced by hypoxia and interleukin lb. J Rheumatol. 1997;24:1253–9. [PubMed] [Google Scholar]
- 23.Fava RA, Olsen N, Keski-Oja J, Moses HL, Pincus T. Active and latent forms of transforming growth factor β activity in synovial effusions. J Exp Med. 1989;169:291–6. doi: 10.1084/jem.169.1.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevens CR, Blake DR, Merry P, Revell PA, Levick JR. A comparative study by morphometry of the microvasculature in normal and rheumatoid synovium. Arthritis Rheum. 1992;34:1508–13. doi: 10.1002/art.1780341206. [DOI] [PubMed] [Google Scholar]
- 25.Connoly DT, Heuvelman DM, Nelson R, Olander JV, Eppley B, Delfino JJ, Siegel NR. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–8. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berse B, Brown LF, Van De Water L, Dvorak HF, Senger DR. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages and tumors. Mol Biol Cell. 1992;3:211–20. doi: 10.1091/mbc.3.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]