

Abstract
To investigate the early events of Helicobacter pylori infection in a mouse model, CD1 mice were infected with a type I (CagA+/VacA+) H. pylori strain. Up to 4 weeks after infection the majority of gastric tissue biopsies were positive in culture. Immunohistochemical analysis showed that inflammatory changes started to occur after 3 weeks. Four weeks after infection a significant increase in T cells was observed in the cardia/corpus region of the stomachs of infected mice. These T cells were CD4+ and CD8+, and they were located in an area with increased expression of MHC class II antigens. In 50% of the infected mice also an increased number of mast cells was seen. Furthermore, aggregates of B and T cells were present in the submucosa. Characterization of cytokines by immunohistochemistry showed an increase in IL-5-secreting cells in the inflamed area of the infected stomach. No difference was observed between interferon-gamma (IFN-γ)-, IL-4- and IL-10-secreting cells in control and infected mice. These results suggest that no polarized T-helper cell response was present at this early phase of infection. Infection with H. pylori also induced a serum response and especially IgG was increased after 4 weeks of infection. However, no particular increase in IgG1, IgG2a or IgG3 isotype was observed. Part of the serum antibodies was directed against lipopolysaccharide (LPS), but no evidence for anti-Lewis antibodies or antibodies against epitopes on the gastric mucosa was found.
Keywords: Helicobacter pylori, gastritis, immune response, serology, animal model
INTRODUCTION
Colonization of the human stomach with Helicobacter pylori is associated with the development of chronic superficial gastritis and peptic ulcer disease [1]. Despite a cellular and humoral immune response against this bacterium, the infection is not cleared. This ongoing state of inflammation predisposes the mucosa to atrophic changes, which might eventually result in the development of carcinoma [2]. An aberrant proliferation of B cells in patients with lymphoma has also been shown to be a result of H. pylori infection [2].
In humans the inflammation is primarily present in the antrum, the distal part of the stomach, where the bacterium is residing. Several studies have shown that not only cells of the specific immune system, like B and T cells, are involved in the inflammatory reaction, but also cells of the innate system such as monocytes and mast cells [3–5]. B cells are present in organized structures like lymphoid follicles with germinal centres, whereas T cells are found more dispersed in the lamina propria. Most of these T cells are CD4+ [3] and recent studies indicate that they show a T-helper 1 (Th1) phenotype [6,7]. These inflammatory cells are only present in areas where epithelial cells show an increased expression of MHC class II molecules [3,5].
These data demonstrate that H. pylori infection induces gastric inflammation but give limited information on the role inflammatory cells play during infection. Helicobacter pylori infection is thought to be acquired during childhood, but gastric disease usually becomes manifest after a prolonged time of infection. Only three cases of acute H. pylori infection have been described and these reports indicate that polymorphonuclear granulocytes (PMN) may also contribute to pathology [1,8,9].
An animal model, in which the infection can be studied in a more controlled manner, would thus be of great help to study the early inflammatory changes. Although H. pylori evokes gastritis in several animal models, the nature of inflammation does not always resemble human disease. In the pig model, for example, the main inflammatory cell is the CD8+ T cell [10], whereas in humans CD4+ T cells are increased. The H. felis mouse model has been shown to mimic many features of human disease, like increased numbers of PMN, T, B and mast cells [11,12], but H. felis differs in several ways from H. pylori. Helicobacter felis does not adhere to epithelial cells and has bipolar flagella, which enable the bacterium to invade the gastric pits more deeply than H. pylori [13]. But more importantly, H. felis does not have a pathogenicity island [12] which contains several genes, among which are a cytotoxin-associated gene A (cagA) and a vacuolating cytotoxin (vacA). The products of these genes have been shown to be associated with more severe inflammation in H. pylori infection [14,15].
For these reasons we have used a mouse model [16] to study the early histopathological changes during infection. In this model, inflammation is more severe in mice infected with a CagA/VacA+H. pylori strain compared with mice, which are infected with a H. pylori strain lacking these antigens. However, the nature of these inflammatory cells is at present unknown. To learn more about the inflammatory cells that are involved in the early phase of gastritis an outbred strain of mice, CD1, was infected with a CagA/VacA+H. pylori strain.
Recently, several investigations have indicated that not only the cellular but also the humoral response might contribute to pathology in H. pylori infection [17,18]. In a large population of patients with H. pylori infection autoantibodies are present which react with epitopes present on the gastric mucosa. Lewis x and y have been suggested to be the target of these autoantibodies [19]. Although in a mouse model anti-Lewis antibodies have been shown to cause gastric pathology [20], in humans Lewis structures do not seem to be the target of the autoantibodies [21]. Only limited information about induction of anti-Lewis antibodies or against other epitopes is available in experimental H. pylori infection. Therefore we investigated whether antibodies against H. pylori were present in the serum during the first 4 weeks of infection and whether these antibodies were directed against epitopes expressed by the gastric mucosa.
MATERIALS AND METHODS
Bacteria
Helicobacter pylori SPM326 (CagA+/VacA+) [16] was cultured on blood agar plates, containing 10% (v/v) horse blood under microaerobic conditions for 3–5 days at 37°C.
Experimental design
Male 6–8-week-old CD1 (SPF) mice (Charles River, Calco, Italy) were housed in conventional conditions and had free access to commercial food and water. Mice (n = 32) were inoculated with bacteria as described previously [16]. In short, 0.2 ml 0.2 m NaHCO3 was given by feeding needle to neutralize the acidity of the stomach, and directly afterwards 2 × 109 colony-forming units (CFU) in 0.1 ml PBS were given by the same route. Control mice (n = 16) received sterile PBS. This procedure was repeated twice at 48-h intervals. At 1, 2, 3 and 4 weeks post-inoculation, eight mice of the infected and four mice of the control group were killed. Their stomachs were resected and divided longitudinally into two halves. One half was rubbed gently over the surface of freshly prepared blood agar plates supplemented with 50 μg/ml amphotericin B, 100 μg/ml vancomycin, 3.3 μg/ml polymyxin B, 200 μg/ml bacitracin, and 10.7 μg/ml nalidixic acid [22]. The blood agar plates were incubated as described above and H. pylori was identified by Gram's stain, and a positive reaction in urease, oxidase and catalase tests. The other half of the stomach was frozen immediately for investigation of microscopic tissue damage and immunohistochemistry. Serum was collected for measurement of H. pylori-specific antibodies, antibodies specific for lipopolysaccharide (LPS), and autoantibodies.
Histopathological evaluation
Serial cryostat sections (8 μm) were picked up on gelatin-coated slides and air-dried. After fixation for 10 min in Baker's formalin (4% formalin, 1% CaCl2 in distilled water) the sections were stained with haematoxylin and eosin (H–E). Longitudinal sections of the stomach that contained mucosal tissue from the non-glandular part of the stomach, the cardia, corpus and antrum were investigated for disruption of the glandular structure and presence of ulcers.
Histochemical staining of mucosal mast cells in situ
Cryosections were fixed for 5 min in Baker's formalin and washed in 50 mm Tris–HCl buffer pH 6.8. The sections were then incubated vertically in chymase reaction mixture for 20 min at 37°C. Chymase reaction mixture contained 0.53 mm naphthol-AS-d-chloroacetate, 0.3 mm fast blue BB salt (Sigma Chemical Co., St Louis, MO), 40 mm NaF (Baker Chemicals, Deventer, The Netherlands) and 1% Triton X-100 (Merck, Darmstadt, Germany). After washing in Tris buffer the sections were counterstained with haematoxylin and mounted in Aquamount (BHH Laboratory Supplies, Poole, UK).
Immunohistochemistry
The cryosections were fixed in pure acetone and a two-step immunoperoxidase staining method was used as described previously [23]. In short, slides were incubated horizontally for 60 min at room temperature with a solution of the first step MoAb in 0.01 m PBS pH 7.4, with 0.5% bovine serum albumin (BSA). The slides were washed three times in PBS and subsequently incubated with the appropriate peroxidase-conjugated antiserum, dilution 1:100 (Dako, Glostrup, Denmark) in PBS with 0.5% BSA and 10% normal mouse serum, for 30 min. After being rinsed in PBS, sections were stained for peroxidase activity with 3,3′-diaminobenzidine-tetra-hydrochloride (Sigma) in 0.05 m Tris–HCl pH 7.6 containing freshly added 0.01% H2O2 [24]. After the slides had been washed in PBS, they were counterstained with haematoxylin, dehydrated and mounted in Entellan (Merck). Control slides were incubated in PBS with 0.5% BSA in the first step, instead of the first specific antibody, and examined for non-specific staining. Sections of the spleen were always included as positive control. Staining with MoAbs RA3-6B2 (B220 on B cells) [25], 59-AD-22 (CD90) [26], MT4 (CD4) [27], Lyt-2 (CD8) [26], MOMA-2 [28] and M5.114 (MHC class II) [29] was carried out on consecutive sections. PMN were detected by staining of endogenous myeloperoxidase.
Immunohistochemical detection of cytokines
Staining of cytokines was performed as described above. Antibodies were directed against the following cytokines: Interferon-gamma (IFN-γ) (XMG1.2) [30], IL-4 (BVD6) [31], IL-5 (TRFK5) [32] and IL-10 (2A5) [33].
Quantification of cells in gastric mucosa
The number of mast cells, PMN, T cells, and cytokine-secreting cells was determined using a microscope with a calibrated ocular grid (0.016 mm2; Zeiss, Weesp, The Netherlands). Cells were counted with object magnification × 40 in the most inflamed part of the stomach. Twenty-four grids divided over three sections were counted for each infected mouse. Then the number of cells/mm2 per mouse was calculated. B cells were only present in lymphoid follicles or aggregates and in some mice these structures showed an unequal distribution over the sections. Therefore, the numbers of B cells were not counted, but the absence or presence of follicles and aggregates was scored per mouse, by giving a score of 0 or 1, respectively. MHC class II expression was graded on a 0–3 scale in the most severely affected × 20 microscopic field, as follows: 0, MHC class II expression as observed in control mice (only a few cells lying in between the glands show MHC class II expression, gland cells show no expression); 1, MHC class II expression by 1/3 of the mucosal gland cells; 2, MHC class II expression by 2/3 of the mucosal gland cells; 3, MHC class II-expressing cells by all gland cells extending from lumen to submucosa.
Measurement of H. pylori-specific serum antibodies
IgM, IgG and IgG1, IgG2a and IgG3 subtype antibodies against H. pylori were measured by an ELISA as described previously [19]. Microtitre plates were coated overnight at room temperature with whole-cell sonicates of H. pylori (0.2 mg protein/well). Plates were washed with PBS containing 0.05% Tween 80 (PBS–T). Sera were diluted 1:100 in PBS–T, subsequently diluted two-fold and incubated overnight at room temperature. After washing the plates, antibodies were detected with peroxidase-labelled goat anti-mouse IgM and IgG (Amarican Qualex, La Miranda, CA) diluted 1:1000 in PBS–T with 0.5% normal goat serum. Incubation of biotinylated rabbit anti-mouse IgG1, IgG2a and IgG3 (Zymed Labs, San Francisco, CA) diluted 1:4000 in PBS–T with 0.5% normal rabbit serum was followed by peroxidase-conjugated streptavidin (Dako) diluted 1:5000 in PBS–T. Colour was developed with H2O2 and OPD in citrate-phosphate buffer pH 5.5 for 30 min at room temperature, and the optical density (OD) was read at 490 nm after stopping the reaction with 50 μl of sulphuric acid. Serum from immunized mice was used as positive control and arbitrarily set at 10 000 U/ml. ODs were converted to arbitrary units/ml after linear regression upon the positive standard.
Measurement of antibodies specific for LPS, H+/K+-ATPase, and Lewis antigens
Antibodies specific for LPS, H+/K+-ATPase, and Lewis antigens were detected by ELISA as described above. In short, 100 μl of pig H+/K+-ATPase, LPS (pooled LPS from LPS serotypes O1 to O6, and from strains P466 and MO19) and synthetic Lewis antigens (all at 1 mg/ml suspended in PBS) were added to the wells of microtitre plates and incubated overnight at room temperature. Plates were washed with PBS–T. Pooled sera from mice infected for 4 weeks and an age-matched control group were diluted 1:100 in PBS–T and incubated overnight at room temperature. Incubation with conjugate and subsequent colour development were done as described above. The following controls were used: binding of mouse sera to PBS- and human serum albumin-coated wells; binding of conjugate to antigen-coated wells. OD values were expressed after subtraction of the background values, which were < 0.5.
Detection of autoantibodies
To determine whether autoantibodies were present in the serum, sections of stomachs of control mice were incubated with pooled serum of infected and control mice. Sera were diluted 1:50 in PBS/BSA solution and the procedure described under immunohistochemistry was followed. Serum from immunized mice was used as positive control.
Statistical analysis
Comparison of the number of inflammatory cells/mm2 between infected and control mice was done by the Mann–Whitney U-test. The presence of B cells in lymphoid follicles and aggregates, MHC class II expression and humoral H. pylori- and antigen-specific responses were evaluated by Student's t-test. A value of P < 0.05 was considered statistically significant.
RESULTS
Bacteria were cultured from the stomach to establish whether mice were colonized by H. pylori. The stomachs of at least six out of eight mice were colonized by H. pylori at 1, 2, 3 and 4 weeks post-inoculation. Analysis of H–E-stained sections of the stomach showed that 3 weeks after infection the number of mononuclear cells started to increase in the cardia/corpus region. No epithelial damage or ulcers were observed during the first 4 weeks of infection. The structure of the glands was slightly distorted in areas with increased numbers of inflammatory cells.
Presence of chymase-positive mast cells
In control mice a few chymase-positive mast cells were always present in the submucosa along the entire stomach, whereas in the lamina propria they were primarily present in the cardia/corpus region. Although 50% of the infected mice showed an increase in mast cells in the mucosa of the cardia/corpus, comparison of mast cell numbers of all eight infected mice with the numbers in the control group did not show a significant increase (P > 0.05) (Fig. 1a).
Fig. 1.
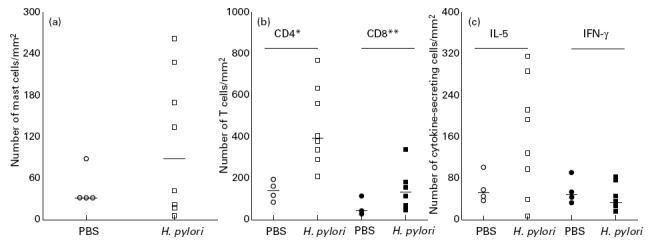
(a) Number of mast cells/mm2 in the cardia of control (○) and infected (□) mice 4 weeks post-inoculation. The increase in mast cells in infected mice was not significant compared with mast cell numbers in control mice (P > 0.05). (b) Number of CD4+ (open symbols) and CD8+ (closed symbols) T cells/mm2 in the cardia/corpus region of control (○,•) and infected (□,▪) mice. Infected mice showed a significant increased number of both CD4+ and CD8+ T cells, although CD4+ cells constituted the major component. Horizontal lines denote median values. *P < 0.004; **P < 0.05. (c) Number of IL-5 (open symbols) and IFN-γ (closed symbols) secreting cells/mm2 in control (○,•) and infected (□,▪) mice. An increase in IL-5-secreting cells was only present in 50% of infected mice and this increase was not significant compared with the control group (P > 0.05). No significant difference was present between the number of IFN-γ-secreting cells in infected and control mice (P > 0.05). Horizontal lines denote median values.
Immunohistochemical analysis of H. pylori-induced gastritis
Immunohistochemical analysis showed that in control mice a few Thy-1+ T cells were present in the mucosa. In infected mice, however, a significant increase in T cells was found co-localized with mast cells in the mucosa and submucosa of the cardia/corpus, whereas the antrum did not show any inflammation (results not shown). Staining with anti-CD4 and anti-CD8 MoAbs showed that both types of T cells were significantly increased, with CD4+ T cells being the most frequent cell type (Fig. 1b). To investigate whether these cells showed a polarized Th response, the numbers of IFN-γ (a Th1 cytokine), IL-4, IL-5 and IL-10 (Th2 cytokines) secreting cells in infected mice were compared with their numbers in control animals. Very few, if any, IFN-γ-, IL-4- and IL-10-secreting cells were present in the cardia or any other part of the stomach of infected as well as control animals. A large variation in the number of IL-5-secreting cells was observed in the infected group, which was not present in the control mice. Comparison of the numbers of IL-5-secreting cells in these two groups showed that the increase was not significant (Fig. 1c).
In infected mice a significant increase in number of follicles and aggregates with B cells was observed compared with control mice (Table 1). The small aggregates and follicles were situated in the submucosa of the cardia and corpus, but were absent in other parts of the stomach.
Table 1.
Presence of B cells in lymphoid aggregates/follicles and MHC class II expression in control and Helicobacter pylori-infected mice at 4 weeks post-inoculation

In infected mice the inflamed gastric tissue showed a significant increase in expression of MHC class II by epithelial gland cells (Table 1), whereas in control mice MHC class II expression was restricted to a few cells that were present between the glands and a few cells which were lining the submucosa.
No increase in MOMA-2+ cells or PMN was detected in any part of the stomach (results not shown).
H. pylori-specific antibodies
Because a T cell-mediated response affects not only the cellular but also the humoral response, we also analysed whether H. pylori-specific antibodies were present in the serum. As the increase in T cells was most evident after 4 weeks of infection, the analysis was restricted to this time point. Figure 2 shows a significant increase in H. pylori-specific antibodies of the IgG isotype, whereas in infected animals the IgM isotype shows only a marginal increase, which is not significant. Further subtyping of the H. pylori-specific IgG serum antibodies did not show a significant increase in either IgG1, IgG2a or IgG3 (results not shown).
Fig. 2.
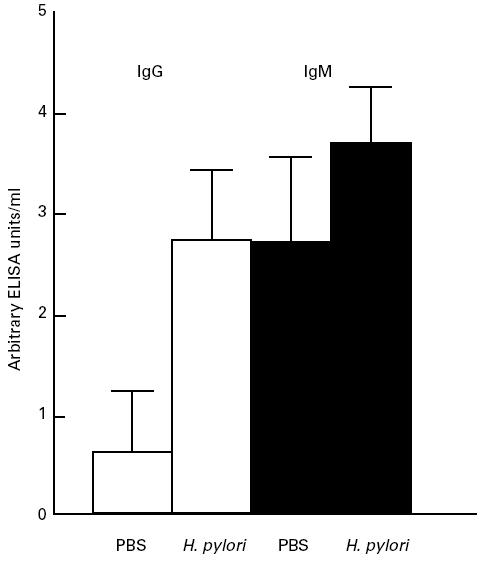
Serological response to sonicated antigens of Helicobacter pylori of control and infected mice as measured in ELISA. The IgG response in infected mice was increased significantly after 4 weeks of infection (P < 0.0001), whereas the IgM response showed only a slight non-significant increase (P > 0.05).
Presence of antibodies directed against LPS and autoantigens
To investigate if antibodies against autoantigens were induced during H. pylori infection, two methods were used: first, pooled sera from mice which were infected for 4 weeks and age-matched controls were tested in ELISA, and second, these sera were tested for the presence of cross-reacting antibodies on normal gastric sections.
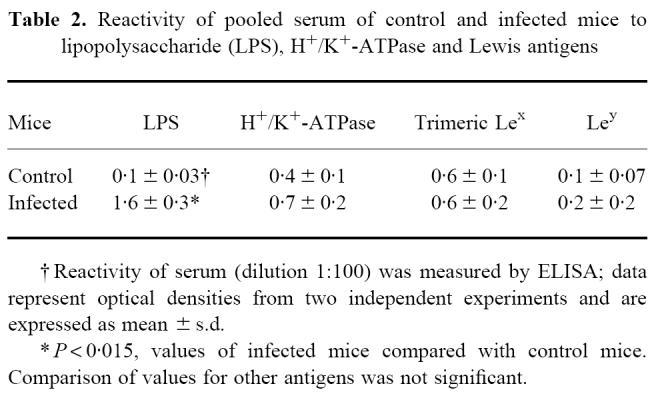
The results of the ELISA are shown in Table 2. The concentration of antibodies against LPS was significantly increased in infected mice, but no difference in reactivity to H+/K+-ATPase, trimeric Lewis x or Lewis y was observed between infected and control mice. Also in sections both sera did not cross-react with gastric tissue (results not shown).
Table 2.
Reactivity of pooled serum of control and infected mice to lipopolysaccharide (LPS), H+/K+-ATPase and Lewis antigens
DISCUSSION
In this study we have shown that 4 weeks after inoculation of CD1 mice with H. pylori a significant increase in T cells, B cells and an increased expression of MHC class II antigens is present in the gastric mucosa. Mast cells showed only a slight but non-significant increase. Immunohistochemical analysis of the cytokines that were secreted in the inflamed area and the distribution of IgG2a or IgG1 isotypes in the serum gave no evidence for a polarized Th response at this time point. An increased response against LPS was present in the serum of infected mice, but no antibodies against Lewis structures or proton pump were detected.
In humans H. pylori infection has a very slow progression and it takes many years before disease symptoms become manifest. Although in vitro gastric epithelial cells are able to respond within hours to adherence of H. pylori [15,34,35], the in vivo situation seems to be more complex. In the present study we showed that in mice the first signs of gastric inflammation occurred 3 weeks after inoculation. Adherence to epithelial cells alone might not be enough for activation of inflammatory cells; moreover, bacterial antigens have to leak through the lamina propria after the bacterium has invaded the tight junctions. It was remarkable that the inflammation in infected mice was only present in the cardia/corpus region of the stomach and not in the antrum. It has been reported that in humans H. pylori is also present in the cardia and causes inflammation at that site [36]. In mice infected with H. felis a similar result has been reported [37].
Mast cells play an important role in the initiation and regulation of inflammation. Nakajima et al. [4] have shown that in humans increased mast cell numbers are associated with gastric inflammation, irrespective of the presence of H. pylori. This indicates that H. pylori may not play a direct role in the activation of mast cells in the human stomach. Nevertheless, mast cells appear to play an important role in pathology, because their numbers are significantly increased in patients with peptic ulcer disease compared with patients with other types of gastric disease [4]. Also, in H. pylori-infected mice they seem to play a role in gastric inflammation, as well as in H. felis infection [12]. The exact role of mast cells in the pathogenesis of gastric disease remains to be established.
Increased MHC class II expression in the stomach may be induced by IFN-γ, which is secreted by activated lymphocytes [3,5]. However, in this study no difference in IFN-γ secretion in control or infected mice was observed, which suggests that in CD1 mice increased expression of MHC class II antigens is induced by different mechanisms. Gastric epithelial cells are generally not considered to function as professional antigen-presenting cells, but data published recently indicate that these cells are capable of expressing the costimulatory molecules CD80 and CD86 [38]. The location of epithelial cells in the stomach enables them to be in contact with luminal antigens as wells as T cells that are present in the lamina propria, and in this way could induce activation of the latter. Although it is unknown if these costimulatory molecules are present on gastric epithelial cells in mice, our finding that MHC class II expression on epithelial cells and the number of CD4+ T cells increased simultaneously, strongly suggests that in mice the sequence of events may resemble that in humans.
The cytokines secreted by these T cells were analysed to obtain information about the type of Th response they displayed. A slight but non-significant increase in IL-5-secreting cells was found in infected mice, whereas the number of IFN-γ-, IL-4- and IL-10-secreting cells was very low in both infected and control animals. Because T cells and mast cells were co-localized, IL-5 production by mast cells must also be taken into account. These results seem to indicate that 4 weeks after infection no distinct Th1 or Th2 response had developed, which was further supported by serology: although a significant increase in H. pylori-specific antibodies was present in the serum, none of the isotypes which are often increased in a Th1 or Th2 response prevailed. Recent data indicate that only after > 10 weeks of infection with H. pylori does IgG2a become the prevailing subtype in serum of mice [39]. This shows that the polarization towards a Th1 response develops slowly in CD1 mice. Several other studies in humans and animals also indicate that CD4+ T cells of the Th1 lineage are the main type of inflammatory cell in Helicobacter gastritis [6,7,40,41]. Adoptive transfer studies of Th1 cells isolated from H. felis-infected mice have recently shown that these cells are able to exacerbate gastric inflammation in recipients and are not able to reduce the bacterial load [40]. However, transfer of Th2 cells isolated from immunized mice resulted in a more effective reduction of bacteria. A Th1 response seems thus to be associated with pathogenesis of disease, whereas a Th2 response seems to be associated with protection or control of infection [42].
The presence of large numbers of CD4+ T cells in an area with increased MHC class II expression occurs in several autoimmune diseases in humans, and has also been reported to occur in an experimental model of autoimmune gastritis in mice [43]. These mice develop an antibody response against gastric H+/K+-ATPase, the proton pump, which is located in parietal cells. Several studies have shown that a large number of patients who are infected with H. pylori have serum antibodies that cross-react with normal gastric mucosa [17–19]. In some patients antibodies react with the canalicular structures of the parietal cell [17,18]. Lewis x and y have been proposed as possible inducers of these autoantibodies, because Lewis antigens are expressed on H. pylori LPS and on the proton pump and gastric mucin in the host [19]. Furthermore, in mice experimentally infected with H. pylori an anti-Lewis x response was present after 8 weeks of infection [19]. As in humans, also in H. pylori-infected mice an anti-LPS antibody response was present in the serum, but no increased concentrations of anti-Lewis x or y antibodies were detected. A possible explanation for this discrepancy might be that the period of infection in our study was not long enough to develop an anti-Lewis response. Recently, it has become clear that infected humans have very low anti-Lewis antibody titres. In addition, absorption of autoimmune sera with H. pylori did not influence autoreactivity of the sera [21]. These results suggest that not Lewis antigens but other epitopes on the gastric proton pump are recognized by the autoantibodies.
Acknowledgments
We would like to thank Dr B. Appelmelk for helpful discussion. We would also like to thank M. Marchetti, T. Verboom and M. Stor for technical assistance.
References
- 1.Marshall B, Armstrong JA, McGechie DB, et al. Attempt to fulfil Koch's postulates for pyloric Campylobacter. Med J Austral. 1985;142:436–9. doi: 10.5694/j.1326-5377.1985.tb113443.x. [DOI] [PubMed] [Google Scholar]
- 2.Blaser MJ, Parsonnet J. Parasitism by the ‘slow’ bacterium Helicobacter pylori leads to altered gastric homeostasis and neoplasia. J Clin Invest. 1994;94:4–8. doi: 10.1172/JCI117336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papadimitriou CS, Ioachim-Velogianni EE, Tsianos EB, et al. Epithelial HLA-DR expression and lymphocyte subsets in gastric mucosa in type B chronic gastritis. Virch Arch A (Pathol Anat) 1988;413:197–204. doi: 10.1007/BF00718611. [DOI] [PubMed] [Google Scholar]
- 4.Nakajima S, Krishnan B, Ota H, et al. Mast cell involvement in gastritis with or without Helicobacter pylori. Gastroenterol. 1997;113:746–54. doi: 10.1016/s0016-5085(97)70167-7. [DOI] [PubMed] [Google Scholar]
- 5.Engstrand L, Scheynius A, Påhlson C, et al. Association of Campylobacter pylori with induced expression of class II transplantation antigens on gastric epithelial cells. Infect Immun. 1989;57:827–32. doi: 10.1128/iai.57.3.827-832.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karttunen R, Karttunen T, Ekre H-PT, MacDonald TT. Interferon gamma and interleukin 4 secreting cells in the gastric antrum in Helicobacter pylori positive and negative gastritis. Gut. 1995;36:341–5. doi: 10.1136/gut.36.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Elios MM, Manghetti M, De Carli M, et al. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J Immunol. 1997;158:962–7. [PubMed] [Google Scholar]
- 8.Morris A, Nicholson G. Ingestion of Campylobacter pyloridis causes gastritis and raised fasting gastric pH. Am J Gastroenterol. 1991;82:192–9. [PubMed] [Google Scholar]
- 9.Sorbala GM, Crabtree JE, Dixon MF, et al. Acute Helicobacter pylori infection: clinical features, local and systemic immune response, gastric mucosal histology, and gastric juice ascorbic acid concentrations. Gut. 1991;32:1415–8. doi: 10.1136/gut.32.11.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krakowka S, Ringler SS, Eaton KA, et al. Manifestations of the local gastric immune response in gnotobiotic piglets infected with Helicobacter pylori. Vet Immunol Immunopathol. 1996;52:159–73. doi: 10.1016/0165-2427(95)05547-9. [DOI] [PubMed] [Google Scholar]
- 11.Fox JG, Blanco M, Murphy JC, et al. Local and systemic immune responses in murine Helicobacter felis active chronic gastritis. Infect Immun. 1993;61:2309–15. doi: 10.1128/iai.61.6.2309-2315.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohammadi M, Redline R, Nedrud J, et al. Role of the host in pathogenesis of Helicobacter-associated gastritis. H. felis infection of inbred and congenic mouse strains. Infect Immun. 1996;64:238–45. doi: 10.1128/iai.64.1.238-245.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee A, Fox JG. Animal models for vaccine development. In: Ernst PB, Michetti P, Smith PD, editors. The immunobiology of H. pylori: from pathogenesis to prevention. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 255–71. [Google Scholar]
- 14.Ghiara P, Marchetti M, Blaser MJ, et al. Role of the Helicobacter pylori virulence factors vacuolating cytotoxin, CagA, and urease in a mouse model of disease. Infect Immun. 1995;63:4154–60. doi: 10.1128/iai.63.10.4154-4160.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crabtree JE, Covacci A, Farmery SM, et al. Helicobacter pylori induced interleukin-8 expression in gastric epithelial cells is associated with CagA positive phenotype. J Clin Pathol. 1995;48:41–45. doi: 10.1136/jcp.48.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marchetti M, Aricò B, Burroni D, Figura N, Rappuoli R, Ghiara P. Development of a mouse model of Helicobacter pylori infection that mimics human disease. Science. 1995;267:1655–8. doi: 10.1126/science.7886456. [DOI] [PubMed] [Google Scholar]
- 17.Faller G, Steininger H, Kränzlein J, et al. Antigastric autoantibodies in Helicobacter pylori infection: implications of histological and clinical parameters of gastritis. Gut. 1997;41:619–23. doi: 10.1136/gut.41.5.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Negrini R, Savio A, Poiesi C, et al. Antigenic mimicry between Helicobacter pylori and gastric mucosa in the pathogenesis of body atrophic gastritis. Gastroenterol. 1996;111:655–65. doi: 10.1053/gast.1996.v111.pm8780570. [DOI] [PubMed] [Google Scholar]
- 19.Appelmelk BJ, Simoons-Smit I, Negrini R, et al. Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharides and host lewis blood group antigens in autoimmunity. Infect Immun. 1996;64:2031–40. doi: 10.1128/iai.64.6.2031-2040.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Negrini R, Lisato L, Zanella I, et al. Helicobacter pylori infection induces antibodies cross-reacting with human gastric mucosa. Gastroenterol. 1991;101:437–45. doi: 10.1016/0016-5085(91)90023-e. [DOI] [PubMed] [Google Scholar]
- 21.Faller G, Steininger H, Appelmelk B, et al. Evidence of novel pathogenic pathways for the formation of antigastric autoantibodies in Helicobacter pylori gastritis. J Clin Pathol. 1998;51:244–5. doi: 10.1136/jcp.51.3.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McColm AA, Bagshaw J, O'Malley C, et al. Development of a mouse model of gastric colonisation with Helicobacter pylori. Gut. 1995;37(Suppl. 1):A50. [Google Scholar]
- 23.Van Doorn NEM, Van Rees EP, Namavar F, et al. Local cellular immune response in the acute phase of gastritis in mice induced chemically and by Helicobacter pylori. J Med Microbiol. 1998;47:863–70. doi: 10.1099/00222615-47-10-863. [DOI] [PubMed] [Google Scholar]
- 24.Graham RC, Karnovsky MC. The early stages of absorption of injected horseradish peroxidase in the proximal tubules of mouse kidney: ultrastructural cytochemistry by a new technique. J Histochem Cytochem. 1966;14:291–302. doi: 10.1177/14.4.291. [DOI] [PubMed] [Google Scholar]
- 25.Coffman RL. Surface antigen expression and immunoglobulin gene rearrangement during mouse pre-B cell development. Immunol Rev. 1985;69:5–28. doi: 10.1111/j.1600-065x.1983.tb00446.x. [DOI] [PubMed] [Google Scholar]
- 26.Ledbetter JA, Herzenberg LA. Xenogeneic monoclonal antibodies to mouse lymphoid differentiation antigens. Immunol Rev. 1979;47:63–90. doi: 10.1111/j.1600-065x.1979.tb00289.x. [DOI] [PubMed] [Google Scholar]
- 27.Pierres A, Naquet P, van Agthoven A, et al. A rat anti-mouse T4 monoclonal antibody H129-10 inhibits proliferation of Ia-reactive T cell clones and delineates two phenotypically distinct (T4+/Lyt2,3− and T4−/Lyt2,3+) subsets among anti-Ia cytotoxic T cell clones. J Immunol. 1984;132:2775–85. [PubMed] [Google Scholar]
- 28.Kraal G, Rep M, Janse M. Macrophages in T and B cell compartments and other tissue macrophages recognized by monoclonal antibody MOMA-2: an immunohistochemical study. Scand J Immunol. 1987;26:653–61. doi: 10.1111/j.1365-3083.1987.tb02301.x. [DOI] [PubMed] [Google Scholar]
- 29.Lemke H, Hämmerling GJ, Hämmerling U. Fine specificity analysis with monoclonal antibodies of antigens controlled by the major histocompatibility complex and by the Qa/TL region in mice. Immunol Rev. 1979;47:175–206. doi: 10.1111/j.1600-065x.1979.tb00293.x. [DOI] [PubMed] [Google Scholar]
- 30.Cherwinski HM, Schumacher JH, Brown KD, Mosmann TR. Two types of mouse helper T cell clones III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. J Exp Med. 1987;166:1229–44. doi: 10.1084/jem.166.5.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abrams JS, Roncarolo MG, Yssel H, et al. Strategies of anti-cytokine monoclonal antibody development: immunoassay of IL-10 and IL-5 in clinical samples. Immunol Rev. 1992;127:5–24. doi: 10.1111/j.1600-065x.1992.tb01406.x. [DOI] [PubMed] [Google Scholar]
- 32.Schumacher JH, O'Garra A, Shrader B, van Kimmenade A, Bond MW, Mosmann TR, Coffman RL. The characterization of four monoclonal antibodies specific for mouse IL-5 and development of mouse and human IL-5 enzyme-linked immunosorbent assays. J Immunol. 1988;141:1576–81. [PubMed] [Google Scholar]
- 33.Chatelain R, Varkila K, Coffman RL. IL-4 induces a Th2 response in Leishmania major-infected mice. J Immunol. 1992;148:1182–7. [PubMed] [Google Scholar]
- 34.Sharma SA, Tummuru MKR, Miller GG, Blaser MJ. Interleukin-8 response and cytokine of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect Immun. 1995;63:1681–7. doi: 10.1128/iai.63.5.1681-1687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smoot DT, Resau JH, Naab T, et al. Adherence of Helicobacter pylori to cultured human gastric epithelial cells. Infect Immun. 1993;61:350–5. doi: 10.1128/iai.61.1.350-355.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Genta RM, Huberman RM, Graham DY. The gastric cardia in Helicobacter pylori infection. Hum Pathol. 1994;25:915–9. doi: 10.1016/0046-8177(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 37.Sakagami T, Dixon M, O'Rourke J, et al. Atrophic gastric changes in both Helicobacter felis and Helicobacter pylori infected mice are host dependent and separate from antral gastritis. Gut. 1996;39:639–48. doi: 10.1136/gut.39.5.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye G, Barrera C, Fan X, et al. Expression of B7-1 and B7-2 costimulatory molecules by human gastric epithelial cells. J Clin Invest. 1997;99:1628–36. doi: 10.1172/JCI119325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghiara P, Rossi M, Marchetti M, et al. Therapeutic intragastric vaccination against Helicobacter pylori in mice eradicates an otherwise chronic infection and confers protection against reinfection. Infect Immun. 1997;65:4996–5002. doi: 10.1128/iai.65.12.4996-5002.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohammadi M, Czinn S, Redline R, et al. Helicobacter-specific cell-mediated immune responses display a predominant Th1 phenotype and promote a delayed-type hypersensitivity response in the stomachs of mice. J Immunol. 1996;156:4729–38. [PubMed] [Google Scholar]
- 41.Bamford KB, Fan X, Crowe SE, et al. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterol. 1998;114:482–92. doi: 10.1016/s0016-5085(98)70531-1. [DOI] [PubMed] [Google Scholar]
- 42.Mohammadi M, Nedrud J, Redline R, et al. Murine CD4 T-cell response to Helicobacter infection: Th1 cells enhance gastritis and Th2 cells reduce bacterial load. Gastroenterol. 1997;113:1848–57. doi: 10.1016/s0016-5085(97)70004-0. [DOI] [PubMed] [Google Scholar]
- 43.Martinelli TM, Van Driel IR, Alderuccio F, et al. Analysis of mononuclear cell infiltrate and cytokine production in murine autoimmune gastritis. Gastroenterol. 1996;110:1791–802. doi: 10.1053/gast.1996.v110.pm8964405. [DOI] [PubMed] [Google Scholar]