

Abstract
Vascular endothelial growth factor (VEGF) is a potent inducer of angiogenesis and is constitutively expressed in the synovium of rheumatoid arthritis (RA). Over-expression of VEGF may play an important role in pathogenic vascularization and synovial hyperplasia of RA. In the present study, we examined whether disease-modifying anti-rheumatic drugs (DMARDs), including bucillamine (BUC), gold sodium thiomalate (GST), methotrexate (MTX) and salazosulfapiridine (SASP), act by inhibiting the production of VEGF by cultured synovial cells of patients with RA. Treatment of cultured synoviocytes with lipopolysaccharide (LPS) significantly increased VEGF production by cultured synovial cells. BUC significantly inhibited LPS-induced VEGF production, while GST tended to inhibit the production of VEGF. The inhibitory effects on VEGF production were dose-dependent. In contrast, MTX and SASP did not affect VEGF production. Reverse transcriptase-polymerase chain reaction (RT-PCR) analysis showed that BUC also inhibited LPS-induced VEGF mRNA expression in RA synovial cells. The present study provides the first evidence that BUC inhibits VEGF production and the expression of its mRNA in synovial cells of RA patients. Our results indicate that the anti-rheumatic effects of BUC are mediated by suppression of angiogenesis and synovial proliferation in the RA synovium through the inhibition of VEGF production by synovial cells.
Keywords: rheumatoid arthritis, synoviocytes, vascular endothelial growth factor, disease-modifying anti-rheumatic drugs, angiogenesis
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory and systemic autoimmune disease characterized by hyperplasia of synovial cells and angiogenesis, which ultimately lead to the destruction of cartilage and bone. Angiogenesis, the growth and proliferation of new blood vessels, is important in a variety of pathophysiologic processes in the RA synovium. A number of angiogenic factors are involved in the neovascularization process in the RA joint. These include acidic fibroblast growth factor (aFGF), basic fibroblast growth factor (bFGF), platelet-derived endothelial cell growth factor and vascular endothelial growth factor (VEGF). These growth factors stimulate vascular endothelial cells in autocrine and paracrine manners [1,2].
VEGF is present in normal tissues [3–6] and certain neoplastic tissues [7–9]. It is a unique peptide growth factor that specifically stimulates the proliferation of endothelial cells [10] and is thought to play an essential role in angiogenesis during a variety of biological processes, including tissue repair and regeneration [11], and tumour growth [8,12,13]. VEGF is also a potent inducer of microvascular extravasation [14,15]. We have previously reported that VEGF polypeptide and mRNA are distributed in the perivascular space and that both are expressed by subsynovial macrophages and synovial lining cells in the synovial tissues of RA patients using immunohistochemical staining, in situ hybridization and reverse transcriptase-polymerase chain reaction (RT-PCR) analysis [16]. In addition, cultured synovial cells are also known to express VEGF under hypoxic conditions or stimulation by IL-1 [17]. Thus, these observations suggest that the constitutive expression of VEGF in rheumatoid synovial cells may play an important role in the pathophysiology of RA synovium.
Several disease-modifying anti-rheumatic drugs (DMARDs) have been used to control RA. While the majority of these DMARDs act as immunomodulatory drugs in RA [18–25], some also act by inhibiting the angiogenic process [26–31]. However, the mechanism of the inhibitory effects of DMARDs on angiogenesis remains obscure.
We speculated that DMARDs inhibit angiogenesis in the synovium of RA by suppressing VEGF production and VEGF mRNA expression in synovial cells. In the present study, we examined the effect of bucillamine (BUC), gold sodium thiomalate (GST), methotrexate (MTX) and salazosulfapiridine (SASP) on the production of VEGF by cultured synovial cells of RA patients.
PATIENTS AND METHODS
Patients and cell preparation
Tissue specimens were obtained from eight patients with RA (stage III or IV) who fulfilled the diagnostic criteria of the American College of Rheumatology with a disease duration of 10–15 years. For comparative analysis, we also obtained tissues from four patients with osteoarthritis (OA). After informed consent, synovial tissue samples were obtained from patients with RA and OA during synovectomy of the knee or total knee joint arthroplasty. The synovial samples were immediately prepared as described previously [32,33]. Briefly, the synovial tissue was cut into small pieces, washed three times in PBS, and treated with 1 mg/ml collagenase (Sigma Chemical Co., St Louis, MO) for 30–60 min at 37°C. The cells were suspended in Ham F-12 medium (Nikken Bio Medical Lab., Kyoto, Japan) containing 10% fetal calf serum (FCS; Flow Labs, McLean, VA), 100 Umg/ml penicillin and 100 μgmg/ml streptomycin. The cell suspension was plated onto 90-mm culture dishes and cultured in a humidified 5% CO2 incubator. When cell cultures reached confluence, synovial cells were treated with trypsin and further passaged to other dishes. The cells used in the present experiments were from passages two to five.
DMARDs
BUC and SA981 (a metabolite of BUC) were obtained from Santen Pharmaceutical Co. (Osaka, Japan). SASP, GST, MTX and dexamethasone (DEX) were obtained from Sigma, Shionogi Co. (Osaka, Japan), Nacalai Tesque (Kyoto, Japan) and Biomal Res. Lab. (Plymouth Meeting, PA), respectively. BUC, GST and SASP were used at concentrations ranging from 1 to 100 μgmg/ml, while those of MTX and DEX ranged from 0.1 to 10 μgmg/ml and 1 ngmg/ml to 1 μgmg/ml, respectively.
These concentrations of DMARDs in vitro were decided according to those in vivo [34–37] and the concentrations in vitro were about from 10–30-fold those in vivo.
Analysis of VEGF concentration in culture supernatants of synovial cells
For the assay of VEGF production, 24-well flat-bottomed microtitre plates were seeded with 5 × 104 cells in the culture medium. After 24 h, cell growth was arrested by replacing the culture medium with 1% FCS in Ham F-12. In the next step, lipopolysaccharide (LPS; Difco Labs, Detroit, MI) with or without DMARDs was added, followed by incubation for another 72 h, and collection of the supernatants at the end of the culture period. The concentration of VEGF in the supernatant was measured by using VEGF ELISA kits (Immuno-Biological Lab Co., Gunma, Japan) and absorbance at 450 nm was recorded using an ELISA plate reader (BioRad, Richmond, CA).
Detection of VEGF mRNA expression by RT-PCR
After washing cultured synovial cells with PBS (−), they were stimulated with 10 μgmg/ml LPS for 24 h, then harvested and used for the extraction of total RNA using Isogen (Nippon Gene, Tokyo, Japan). Poly A mRNA was isolated using a poly AT tract mRNA isolating kit (Promega, Madison, WI). The first strands of cDNA were synthesized from 2 μg of polyadenylated RNA using the First Strand cDNA synthesis kit (Boehringer, Mannheim, Germany). Oligonucleotide primers were produced by a DNA synthesiser according to the human VEGF cDNA sequence (GeneBank no. M32977]: primer 1, 72–92 (5′-TCTTGGGTGCATTGGAGCCTC-3′); and primer 2, 421−401 (5′-AAGCTCATCTCTCCTATGTGC-3′). RT-PCR was performed using a previously described technique [33] with some modifications. For this purpose, 50-μl aliquots of the reaction mixture were prepared, containing 5 μl of 10 × PCR buffer (Takara, Shiga, Japan), 300 ng of template cDNA, and 100 pmol of each of 5′ and 3′ primers. PCR assays for VEGF and β-actin were performed in a thermal cycler (Perkin-Elmer Cetus, Norwalk, CT) for 30 cycles, each consisting of 1 min at 94°C, 1 min at 60°C and 1 min at 72°C. The PCR products were analysed on a 1.5% agarose gel and the density of each band was analysed semiquantitatively using a densitometer.
Proliferation assay by MTS
MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] assay kits (Promega) was used as non-radioactive cell proliferation assays. For this purpose, synovial cells were plated at 1 × 104/well onto 96-well plates (third passage) and incubated for 24 h in Ham F-12 medium containing 10% FCS (Flow Labs) at 37°C in a humidified 5% CO2 atmosphere. The culture media were replaced with F-12 medium containing 1% FCS and incubated for 72 h at 37°C. Following the addition of 20 μl/well of a freshly prepared MTS/PMS solution (100 μl of PMS solution was combined with 2.0 ml of MTS solution immediately before addition to the culture plate containing the cells), the plate was incubated for 4 h at 37°C in a humidified 5% CO2 atmosphere. This was followed by measurement of absorbance at 490 nm using an ELISA plate reader (BioRad).
Statistical analysis
All values were expressed as mean ± s.e.m. Differences between groups were tested for statistical significance using Student's t-test and Sheffe's test. P < 0.05 denoted the presence of a significant difference.
RESULTS
Inhibition of VEGF production in the culture supernatant
LPS, as well as a variety of other agents, are potent stimuli for IL-1β, IL-6 and tumour necrosis factor-alpha (TNF-α) release by synovial cells [38,39]. We first examined whether LPS activates the production of VEGF on synovial cells of patients with RA and OA. After culture of synovial cells in 1% FCS in Ham F-12, we added 10–100 μg/ml LPS and incubated the cells for 72 h. The concentration of VEGF in the supernatants of synovial cell cultures was measured using VEGF ELISA kits. LPS induced the production of VEGF in synovial cells in a dose-dependent manner (data not shown). As shown in Fig. 1, 10 μg/ml LPS caused a two-fold increase in the production of VEGF by synovial cells (Fig. 1). LPS-enhanced production of VEGF was time-dependent, extending between 4 h and 72 h.
Fig. 1.
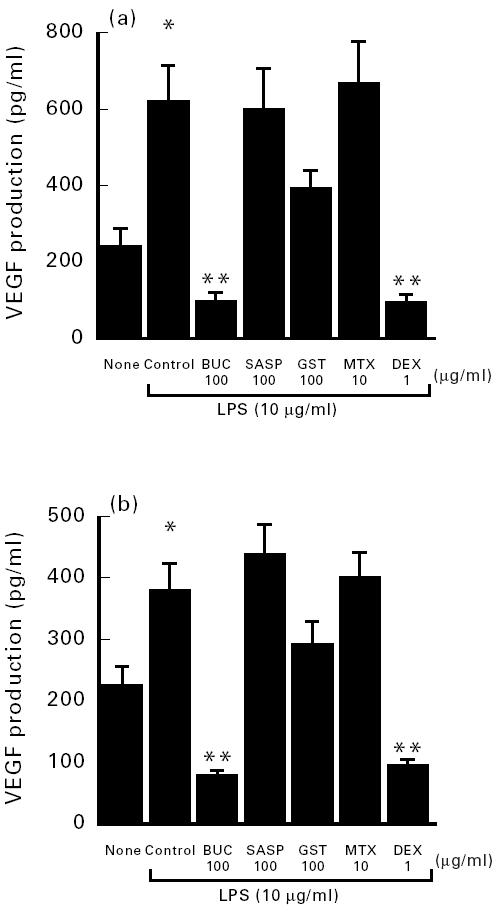
Effect of disease-modifying anti-rheumatic drugs (DMARDs) on vascular endothelial growth factor (VEGF) production by lipopolysaccharide (LPS)-stimulated synovial cells from four patients with osteoarthritis (OA) (a) and eight patients with rheumatoid arthritis (RA) (b). Results represent the mean ± s.e.m. *P < 0.05 compared with unstimulated untreated synoviocytes (none); **P < 0.01 compared with control synoviocytes. BUC, Bucillamine; SASP, salazosulfapyridine; GST, gold sodium thiomalate; MTX, methotrexate; DEX, dexamethasone.
Next, we examined the effects of DMARDs on LPS-induced production of VEGF by synovial cells. After culture of synovial cell in 1% FCS in Ham F-12 and 10 μg/ml LPS, variable concentrations of DMARDs were added, followed by incubation of the cells for another 72 h. LPS-stimulated high VEGF production by cultured synovial cells of patients with RA was inhibited by BUC and DEX, to 78.0 ± 8.2 pg/ml and 93.0 ± 11.2 pg/ml, respectively (Fig. 1). Furthermore, BUC and DEX also inhibited LPS-stimulated VEGF production by OA synoviocytes to 98.8 ± 20.5 pg/ml and 97.0 ± 18.2 pg/ml, respectively (Fig. 1). These values were significantly different from those of DMARD-untreated stimulated synovial cells (380.7 ± 43.2 pg/ml). GST tended to inhibit the production of VEGF by cultured synovial cells, albeit insignificantly. In contrast, MTX and SASP did not inhibit VEGF production by cultured synovial cells of RA patients.
In the next step, we investigated the dose response to BUC, MTX and DEX. As shown in Fig. 2, BUC and DEX inhibited VEGF production in a concentration-dependent manner. BUC significantly inhibited VEGF production from 419.6 ± 17.2 pg/ml (stimulated by LPS 10 μg/ml) to 203.2 ± 14.2 pg/ml when used at a concentration of 30 μg/ml. Similarly, DEX significantly inhibited VEGF production to 156.58 ± 2.47 pg/ml when used at a dose of 0.01 μg/ml.
Fig. 2.

Inhibitory effect of bucillamine (BUC), methotrexate (MTX) and dexamethasone (DEX) on vascular endothelial growth factor (VEGF) production by lipopolysaccharide (LPS)-stimulated synovial cells. Results represent the mean ± s.e.m. of eight patients with rheumatoid arthritis (RA). *P < 0.05 compared with unstimulated untreated synoviocytes (none); **P < 0.01 compared with control synoviocytes.
BUC is a thiol compound that contains two free sulfhydryl groups and differs from d-penicillamine (d-Pen). It has been reported that SA981, a metabolite of BUC with an intramolecular disulphide, has greater effects on B and T cell function than BUC itself [18,41]. In the next step, we investigated whether other thiol compounds such as SA981 and d-Pen exert an inhibitory effect on VEGF production by LPS-stimulated cultured synovial cells. Both BUC (93.3 ± 12.3 pg/ml) and SA981 (169.8 ± 19.4 pg/ml) significantly inhibited VEGF production compared with control cultured synovial cells (466.0 ± 73.3 pg/ml). Furthermore, these effects were concentration-dependent. In contrast, d-Pen did not influence VEGF production (data not shown).
RT-PCR assay of cultured synovial cells
We also examined the effect of DMARDs on VEGF mRNA expression on LPS-stimulated cultured synovial cells using RT-PCR. After treatment for 24 h, total RNA was extracted from the synovial cells and converted to cDNA by RT. A semiquantitative RT-PCR assay was used for the detection of VEGF mRNA, relative to β-actin mRNA expression, as described previously [33]. As shown in Fig. 3, over-expression of VEGF mRNA was detected in LPS-stimulated synovial cells. BUC and DEX, but not MTX and SASP, inhibited VEGF mRNA expression.
Fig. 3.

Reverse transcriptase-polymerase chain reaction (RT-PCR) analysis of vascular endothelial growth factor (VEGF) and β-actin. Note that bucillamine (BUC) as well as dexamethasone (DEX) showed an inhibitory effect on VEGF mRNA.
MTS assay of cultured synovial cells
To investigate whether DMARDs suppressed VEGF production by inhibition of proliferation of cultured synovial cells, we used the MTS assay to examine the effect of DMARDs on the proliferation of these cells. As shown in Fig. 4, all DMARDs tested in the present study, with the exception of SASP, had no effects on synovial cell proliferation. Thus, the inhibitory effects of DMARDs on VEGF production noted above were not due to inhibition of the proliferation of synovial cells, but rather due to inhibition of VEGF polypeptide or VEGF mRNA.
Fig. 4.

Effect of disease-modifying anti-rheumatic drugs (DMARDs) on the proliferation of lipopolysaccharide (LPS)-stimulated synovial cells. Results represent the mean ± s.e.m. of eight patients with rheumatoid arthritis. **P < 0.01 compared with control synoviocytes.
DISCUSSION
The major findings of the present study were the inhibitory effects of DMARDs on VEGF production in both RA and OA. Furthermore, BUC and DEX exerted significant inhibitory effects on VEGF production in a dose-dependent manner. These inhibitory effects were not due to cytotoxic effects or inhibition of proliferation of synovial cells, but rather involved the production or mRNA expression of VEGF. Our results also showed that MTX and SASP had no inhibitory effects on VEGF production even when used at 10-fold concentrations. On the other hand, only a marginal inhibitory effect was noted for GST with regard to the production of VEGF by cultured synovial cells. Our results showed a superior effect for BUC than other DMARDs, and thus it may be the most useful DMARD for inhibition of VEGF production in vivo. It has been reported that the maximal plasma concentration of BUC is 10−5–10−6 m after oral administration of 300 mg BUC, and the concentration in tissues, such as synovium, joint cartilage and other collagen-rich tissues, is higher than that in plasma [40]. It is suggested that the inhibition of VEGF production by BUC in this study possibly occurs in the synovium of treated patients.
BUC is a thiol compound that contains two free sulfhydryl groups, thus making it different from d-Pen. It has been reported that SA981, a metabolite of BUC with an intramolecular disulphide, has greater effects on B cell and T cell function than BUC itself [18,41]. Therefore, we examined the effects of thiol compounds such as SA981 and d-Pen on VEGF production by cultured synovial cells. Our results showed that both BUC and SA981, but not d-Pen, significantly inhibited VEGF production by cultured synoviocytes in a concentration-dependent manner (data not shown). Matsubara et al. [28,31] reported that d-Pen inhibited IL-1-induced synovial and endothelial cell proliferation in the presence of copper sulphate. This observation indicates that hydrogen peroxide produced by d-Pen together with copper inhibits angiogenesis in vivo, and IL-1-induced synovial and endothelial cell proliferation. Therefore, we investigated whether hydrogen peroxide by BUC and SA981 may play a central role in inhibition of VEGF production by thiol compounds. However, the inhibitory effect of BUC did not affect the production of VEGF with or without catalase (data not shown). Thus, these findings suggest the inhibitory effects of BUC and SA981 in synovial cells are not due to the production of hydrogen peroxide.
VEGF is composed of four polypeptides, VEGF121, VEGF165, VEGF189 and VEGF206, produced by alternative splicing of VEGF mRNA. These polypeptides possess a signal peptide composed of 26 amino acids. In particular, VEGF165 and VEGF121 exert dose-dependent mitogenic effects on vascular endothelial cells [6]. On the other hand, despite the presence of a signal peptide, the 24 amino acid insertion common to VEGF189 and VEGF206 apparently prevents effective secretion of these polypeptides. Therefore, VEGF165 and VEGF121 are considered to be present mainly in the culture supernatant, while VEGF189 and VEGF206 are present mainly in cell lysates. Our results show that BUC and DEX inhibited the production of VEGF polypeptide by cultured synovial cells and synovial cell lysates. On the other hand, DEX inhibited VEGF production in rat glioma [42] and similar results were also demonstrated in cultured synovial cells. DEX also showed a significant inhibitory effect at a concentration exceeding that in vivo [43,44]. In this regard, it has also been reported that DEX down-regulates the induced expression of VEGF gene, suggesting a transcriptional repression mechanism within the promotor of the VEGF gene [45]. These findings suggest that VEGF antagonists, that prevent the interaction between VEGF and its receptor, may be beneficial in RA. Further studies are required to understand the mechanisms of the inhibitory effects of BUC on VEGF mRNA expression and production.
Acknowledgments
This work was supported by Grant-in-Aid for Scientific Research, The Ministry of Education, Science, Sports and Culture.
REFERENCES
- 1.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–4. [PubMed] [Google Scholar]
- 2.Leung DW, Cachianes G, Kuang W-J, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–9. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 3.Olson TA, Mohanraj D, Carson LF, Ramakrishnan S. Vascular permeability factor gene expression in normal and neoplastic human ovaries. Cancer Res. 1994;54:276–80. [PubMed] [Google Scholar]
- 4.Sharkey AM, Charnock-Jones DS, Boocock CA, Brown KD, Smith SK. Expression of mRNA for vascular endothelial growth factor in human placenta. J Reprod Fertil. 1993;99:609–15. doi: 10.1530/jrf.0.0990609. [DOI] [PubMed] [Google Scholar]
- 5.Brown LF, Berse B, Tognazzi K. Vascular permeability factor mRNA and protein expression in human kidney. Kidney Int. 1992;42:1457–61. doi: 10.1038/ki.1992.441. [DOI] [PubMed] [Google Scholar]
- 6.Houck KA, Ferrara N, Winer J, Cachianes GI, Li B, Leung DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol. 1991;5:1806–14. doi: 10.1210/mend-5-12-1806. [DOI] [PubMed] [Google Scholar]
- 7.Brown LF, Berse B, Jackman RW, Tognazzi K, Manseau EJ, Senger DR, Dvorak HF. Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in adenocarcinomas of the gastrointestinal tract. Cancer Res. 1993;53:4727–35. [PubMed] [Google Scholar]
- 8.Morii K, Tanaka R, Washiyama K, Kumanishi T, Kuwano R. Expression of vascular endothelial growth factor in capillary hemangioblastoma. Biochem Biophys Res Commun. 1993;194:749–55. doi: 10.1006/bbrc.1993.1885. [DOI] [PubMed] [Google Scholar]
- 9.Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359:845–8. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161:851–8. doi: 10.1016/0006-291x(89)92678-8. [DOI] [PubMed] [Google Scholar]
- 11.Shweiki D, Itin A, Neufeld G, Gitay-Goren H, Keshet E. Patterns of expression of vascular endothelial growth factor (VEGF) and VEGF receptors in mice suggest a role in hormonally regulated angiogenesis. J Clin Invest. 1993;91:2235–43. doi: 10.1172/JCI116450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–5. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 13.Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–7. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 14.McClure N, Healy DL, Robertoson DM. Vascular endothelial growth factor as capillary permeability agent in ovarian hyperstimulation syndrome. Lancet. 1994;344:235–6. doi: 10.1016/s0140-6736(94)93001-5. [DOI] [PubMed] [Google Scholar]
- 15.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–5. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 16.Nagashima M, Yoshino S, Ishiwata T, Asano G. Role of vascular endothelial growth factor in angiogenesis of rheumatoid arthritis. J Rheumatol. 1995;22:1624–30. [PubMed] [Google Scholar]
- 17.Jackson JR, Minton JAL, Ho ML, Wei N, Winkler JD. Expression of vascular endothelial growth factor in synovial fibroblasts is induced by hypoxia and interleukin 1 β. J Rheumatol. 1997;24:1253–9. [PubMed] [Google Scholar]
- 18.Hashimoto K, Lipsky PE. Immunosuppression by the disease modifying antirheumatic drug bucillamine: inhibition of human T lymphocyte function by bucillamine and its metabolites. J Rheumatol. 1993;20:953–7. [PubMed] [Google Scholar]
- 19.Sanders KM, Carlson PL, Littman BH. Effects of gold sodium thiomalate on interferon stimulation of C2 synthesis and HLA-DR expression by human monocytes. Arthritis Rheum. 1987;30:1032–9. doi: 10.1002/art.1780300910. [DOI] [PubMed] [Google Scholar]
- 20.Gubner R, August S, Ginsberg V. Therapeutic suppression of tissue reactivity. II. Effect of aminopterin in rheumatoid arthritis and psoriasis. Am J Med Sci. 1951;221:176–82. [PubMed] [Google Scholar]
- 21.Brody M, Bohm I, Bauer R. Mechanism of action of methotrexate: experimental evidence that methotrexate blocks the binding of interleukin-1β to the interleukin 1 receptor on target cells. Eur J Clin Chem Clin Biochem. 1993;31:667–74. doi: 10.1515/cclm.1993.31.10.667. [DOI] [PubMed] [Google Scholar]
- 22.Seitz M, Dewald B, Ceska M, Gerber S, Baggiolini M. Interleukin-8 in inflammatory rheumatic diseases: synovial fluid levels, relation to rheumatoid factors, production by mononuclear cells, and effects of gold sodium thiomalate and methotrexate. Rheumatol Int. 1992;12:159–64. doi: 10.1007/BF00274936. [DOI] [PubMed] [Google Scholar]
- 23.Seitz M, Loetscher P, Dewald B, Towbin H, Rordorf C, Gallati H, Baggiolini M, Gerber NJ. Methotrexate action in rheumatoid arthritis: stimulation of cytokine inhibitor and inhibition of chemokine production by peripheral blood mononuclear cells. Br J Rheumatol. 1993;34:602–9. doi: 10.1093/rheumatology/34.7.602. [DOI] [PubMed] [Google Scholar]
- 24.Carlin G, Djursäter R, Smedegård G. Sulfasalazine inhibition of human granulocyte activation by inhibition of second messenger compounds. Ann Rheum Dis. 1992;51:1230–6. doi: 10.1136/ard.51.11.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlin G, Djursäter R, Smedegård G. Inhibitory effects of sulfasalazine and related compounds on superoxide production by human polymorphonuclear lymphocytes. Pharmacol Toxicol. 1989;65:121–7. doi: 10.1111/j.1600-0773.1989.tb01141.x. [DOI] [PubMed] [Google Scholar]
- 26.Koch AE, Cho M, Burrows J, Leibovich SJ, Polverini PJ. Inhibition of production of macrophage-derived angiogenic activity by the anti-rheumatic agents gold sodium thiomalate and auranofin. Biochem Biophys Res Commun. 1988;154:205–12. doi: 10.1016/0006-291x(88)90671-7. [DOI] [PubMed] [Google Scholar]
- 27.Kouda M, Yoshino S, Nakamura H, Asano G. Effects of disease modifying antirheumatic drugs (DMARDs) and DEX on IL-1β and IL-6 production by IL-1β-stimulated synovial culture cells. J Nippon Med Sci. 1996;63:1–5. doi: 10.1272/jnms1923.63.419. [DOI] [PubMed] [Google Scholar]
- 28.Matsubara T, Ziff M. Inhibition of human endothelial cell proliferation by gold compounds. J Clin Invest. 1987;79:1440–6. doi: 10.1172/JCI112972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirata S, Matsubara T, Saura R, Tateishi H, Hirohata K. Inhibition of in vitro vascular endothelial cell proliferation and in vivo neovascularization by low dose methotrexate. Arthritis Rheum. 1989;32:1065–73. doi: 10.1002/anr.1780320903. [DOI] [PubMed] [Google Scholar]
- 30.Grootveld M, Blake DR, Sahinoglu T. Control of oxidative damage in rheumatoid arthritis by gold(I)-thiomalate drugs. Free Rad Res Commun. 1990;10:199–200. doi: 10.3109/10715769009149889. [DOI] [PubMed] [Google Scholar]
- 31.Matsubara T, Saura R, Hirohata K, Ziff M. Inhibition of human endothelial cell proliferation in vitro and neovascularization in vivo by d-penicillamine. J Clin Invest. 1989;83:158–67. doi: 10.1172/JCI113853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goto M, Sasano M, Yamanaka H, Miyasaka N, Kamatani N, Inoue K, Nishioka K, Miyamoto T. Spontaneous production of an interleukin 1-like factor by cloned rheumatoid synovial cells in long-term culture. J Clin Invest. 1987;80:786–96. doi: 10.1172/JCI113135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aono H, Hasunuma T, Fujisawa K, Nakajima T, Yamamoto K, Mita S, Nishioka K. Direct suppression of human synovial cell proliferation in vitro by salazosulfapyridine and bucillamine. J Rheumatol. 1996;23:65–70. [PubMed] [Google Scholar]
- 34.Sugawara S, Ishigami M, Kageyama T. Phase 1 study of N-(2-mercapto-2-methylpropionyl)-l-cysteine (SA96). (1) Single administration study. Jpn J Clin Pharmacol Ther. 1985;16:143–52. [Google Scholar]
- 35.Massarella JW, Waller ES, Crout JE, Yakatan GJ. The pharmacokinetics of intramuscular gold sodium thiomalate in normal volunteers. Biopharmaceutics Drug Disposition. 1983;5:101–7. doi: 10.1002/bdd.2510050203. [DOI] [PubMed] [Google Scholar]
- 36.Uchida E, Kai K, Kobayashi S, Oguchi K, Miyazaki Y, Yasuhara H. A study of pharmacokinetics and safety of salazosulfapyridine enteric coated tablet (PJ-306) in healthy Japanese subjects. Jpn J Clin Pharmacol Ther. 1990;21:377–89. [Google Scholar]
- 37.Furst DE. Clinical pharmacology of very low dose methotrexate for use in rheumatoid arthritis. J Rheumatol (Suppl) 1985;12:11–14. [PubMed] [Google Scholar]
- 38.Kageyama Y, Koida Y, Miyamoto S, Yoshida TO, Inoue T. Leukotriene B4-induced interleukin-1β in synovial cells from patients with rheumatoid arthritis. Scand J Rheumatol. 1994;23:148–50. doi: 10.3109/03009749409103049. [DOI] [PubMed] [Google Scholar]
- 39.Falus A, Lakatos T, Smolen J. Dissimilar biosynthesis of interleukin-6 by different areas of synovial membrane of patients with rheumatoid arthritis and osteoarthritis. Scand J Rheumatol. 1992;21:116–9. doi: 10.3109/03009749209095081. [DOI] [PubMed] [Google Scholar]
- 40.Mita S, Matsunaga K. Differences in the effects of the antirheumatic drugs, bucillamine and d-penicillamine, on mitogen-induced proliferation of mouse spleen cells. Agents Action. 1990;30:363–8. doi: 10.1007/BF01966300. [DOI] [PubMed] [Google Scholar]
- 41.Hirohata S, Lipsky PE. Regulation of B cell function by bucillamine, a novel disease-modifying antirheumatic drug. Clin Immunol Immunopathol. 1993;66:43–51. doi: 10.1006/clin.1993.1006. [DOI] [PubMed] [Google Scholar]
- 42.Heiss JD, Papavassiliou E, Merrill MJ, Nieman L, Knightly JJ, Walbridge S, Edwards NA, Oldfield EH. Mechanism of dexamethazone suppression of brain tumor-associated vascular permeability in rats. J Clin Invest. 1996;98:1400–8. doi: 10.1172/JCI118927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kern JA, Lamb RJ, Reed JC, Daniele RP, Nowell PC. Dexamethasone inhibition of interleukin 1 beta production by human monocytes. J Clin Invest. 1988;81:237–44. doi: 10.1172/JCI113301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohase M, Henriken-Destefano D, Sehgal PB. Dexamethasone inhibits feedback regulation of the mitogenic activity of tumour necrosis factor, interleukin-1 and epidermal growth factor in human fibroblasts. J Cell Physiol. 1987;132:271–8. doi: 10.1002/jcp.1041320211. [DOI] [PubMed] [Google Scholar]
- 45.Nauck M, Roth M, Tamm M, Eickelberg O, Wieland H, Stulz P, Perruchoud AP. Induction of vascular endothelial growth factor by platelet-activating factor and platelet-derived growth factor is downregulated by corticosteroids. Am J Respir Cell Mol Biol. 1997;16:398–406. doi: 10.1165/ajrcmb.16.4.9115750. [DOI] [PubMed] [Google Scholar]