

Abstract
The role of soluble receptors for TNF-α (sTNF-Rs) as markers of virus-induced host responses was studied by the use of murine model infections. A marked elevation in serum levels of sTNF-R75, but not sTNF-R55, was found 1 day after infection with vesicular stomatitis virus (VSV). In mice infected with lymphocytic choriomeningitis virus (LCMV), an early increase was also revealed, but peak levels of sTNF-R75 were observed later temporally related to maximal T cell-mediated anti-viral activity. Analysing different well characterized knockout mice, it was found that elevated release of sTNF-R75 into serum early after VSV infection was independent of T cells, whereas interferon (IFN)-α/β seemed to be a major mediator. In contrast, increased release of sTNF-R75 into serum 8 days post-LCMV infection was mediated via T cells but independently of both CD40 ligand and IFN-γ. A simple correlation between release of sTNF-Rs in vivo and macrophage activation in vitro was not present. These findings indicate that sTNF-R75 is indeed a sensitive marker of both innate and specific cell-mediated host reactivity during viral infection, but it is not correlated to a single immunological parameter.
Keywords: soluble TNF receptor I and II in serum, viral infection, innate immunity, T cell-mediated immunity, IFN-α/β
INTRODUCTION
TNF-α is a potent mediator of inflammatory responses produced mainly by activated monocytes/macrophages and T cells. Its biological functions are mediated through two high-affinity cell surface receptors; type I (TNF-R55) of 55–60 kD, and type II (TNF-R75) of 75–80 kD. Most TNF-mediated biological effects have been ascribed to interaction with TNF-R55 [1,2], whereas the role of TNF-R75 has been more elusive, and it has even been suggested that this receptor may exert an attenuating function in certain situations [3]. In accordance with this, TNF-R55 has been found to be of primary importance for the toxic effect of TNF-α, e.g. mice deficient in TNF-R55 are relatively resistant to the development of shock following injection of lipopolysaccharide (LPS) and d-galactosamine [1,2]. However, signalling through TNF-R75 appears to be important for induction of apoptosis in mature T lymphocytes, and TNF-R75-deficient mice are less susceptible to injection of TNF-α [4]. Most cell types and tissues express both types of receptors, and this expression is inducible by interferons, of which interferon-gamma (IFN-γ) is most important [5,6]. Induction of TNF-α production is generally associated with shedding of the extracellular domain of the receptors via proteolytic cleavage. Soluble forms of both TNF receptor types are present in the circulation of normal individuals and have been found to be increased during diseases (e.g. HIV infection, malignant diseases and sepsis) [7–11]. Neutrophils, activated T cells and monocytes seem to be the main sources of sTNF-Rs, but little is known with certainty [12]. The soluble forms of TNF-Rs have retained the capacity to bind TNF-α, and very diverse and often contradictory functions of sTNF-Rs have been suggested [12]. First, sTNF-Rs may serve as decoy receptors and thus reduce the toxic effects of TNF through the binding of bioactive TNF. Binding of TNF could also serve to prolong the circulating half-life, providing a reservoir of bioactive TNF. Finally, soluble receptors could serve as carriers of TNF, and thus contribute to the clearance of TNF. From a more practical viewpoint, analysis of sTNF-R levels is often used to monitor immune activation, and especially sTNF-R75 is taken to be a predictor of a Th1 response in human studies [12].
It was the purpose of this study to evaluate in an animal model whether sTNF-R levels in serum could be used to monitor virus-induced host responses, and—using this model—to try to identify the mechanism underlying any changes observed. It was of particular interest to determine whether sTNF-R levels in serum could be used as a simple and easily accessible parameter of cell-mediated immunity. Systemic viral infections, e.g. measles, HIV or infectious mononucleosis, tend to be characterized by extensive activation of T cells and production of IFN-γ by the T cells [13–18], and from previous studies we know that IFN-γ released in the context of a systemic viral infection causes macrophage activation that sensitizes mice to the lethal effects of LPS [19]. Consequently, we also wanted to investigate whether any increase in sTNF-Rs in serum would correlate with macrophage activation, measured as production of TNF-α and nitric oxide (NO) by splenic adherent cells in vitro. Additionally, we wanted to test whether sTNF-R levels in serum could be used as a predictor of increased susceptibility to septic shock. Systemic infection with lymphocytic choriomeningitis virus (LCMV) was selected as our primary virus model based on the following considerations: murine LCMV infection resembles systemic viral infection in humans such as measles, HIV, and infectious mononucleosis [17,20–23]. Additionally, it is a non-cytopathic virus, causing little or no disease when inoculated by a peripheral route [24,25]. Therefore any effect observed is likely to reflect an altered host response. Besides LCMV, vesicular stomatitis virus (VSV) was used in some experiments. VSV is not a natural mouse pathogen and viral replication is limited [26]. Infection with VSV causes a limited cell-mediated immune response [27]. However, a more distinct innate immune response is observed during this infection compared with the LCMV infection [26,28].
MATERIALS AND METHODS
Mice
C57Bl/6 (B6), C57Bl/6 nu/nu and 129Sv mice were obtained from Bomholtgaard (Ry, Denmark). CD40 ligand-deficient (CD40L −/−) mice (C57Bl/6,129-Cd40l < tm1Imx >) and IFN-γ-deficient (IFN-γ −/−) mice (C57Bl/6-ifg < tm1 >) were the progeny of breeder pairs obtained from The Jackson Laboratory (Bar Harbor, ME). Type I IFN receptor knockout mice (IFN-α/βR −/−, A129) were the progeny of breeder pairs obtained from B&K Universal Ltd (Hull, UK). Female mice, 7–10 weeks old, were used in all experiments, and the mice were always allowed to acclimatize to the local environment for at least 1 week before use. Animals were housed under controlled conditions (SPF) that included testing of sentinels for unwanted infections; no such infections were detected.
Virus
LCMV of the Traub strain, produced and stored as previously described [29], was used in most experiments. Mice to be infected received a dose of 103 LD50 in an i.v. injection of 0.3 ml. Infection by this route is followed by a transient, immunizing infection [24,25].
VSV of the Indiana strain (originally provided by K. Berg of this institute) was used in some experiments. Mice were infected with 106 plaque-forming units (PFU) in an i.v. injection of 0.3 ml; this dose does not cause any disease in immunocompetent mice, but leads to a fatal rabies-like disease in B cell-deficient mice [30].
LPS
LPS from Escherichia coli serotype 055:B5 (Sigma, St Louis, MO) was used.
Polyinosinic-polycytidylic acid
Mice were injected intraperitoneally with 150 μg of polyinosinic-polycytidylic acid (Poly I:C; Sigma) dissolved in 150 μl PBS.
Preparation of splenic macrophages for determination of NO and TNF-α production
Spleens from mice were aseptically removed and transferred to Hanks' balanced salt solution (HBSS). Single-cell suspensions were obtained by pressing the organ through a fine sterile steel mesh, and erythrocytes were lysed by 0.83% NH4Cl treatment. The cells were washed twice with HBSS, and adjusted to 10 × 106 cells/ml in RPMI 1640 containing 10% fetal calf serum (FCS), supplemented with 2-mercaptoethanol (2-ME), l-glutamine, and penicillin–streptomycin solution. Aliquots of 100 μl of cell suspension were added to flat-bottomed 96-well plates (Nunc, Roskilde, Denmark), and splenic macrophages were allowed to adhere for 2 h at 37°C in 5% CO2. Non-adherent cells were removed by washing the wells twice with culture medium. Adherent cells were stimulated with LPS (20 μg/ml, 100 μl), and supernatants were harvested after 2 h for TNF-α measurement and after 48 hr for NO measurement.
Analysis of nitrite concentration in cell culture supernatants
NO production was determined by quantifying nitrite (NO2−), a stable end-product of NO, using the Griess reaction [31]. Briefly, 50 μl of 1% sulfanil-amid/2.5% H3PO4 were added to 50 μl of culture supernatant, and the mixture was incubated for 10 min at room temperature. Absorbance at 540 nm was determined using an ELISA reader, and NO2− concentration was calculated by comparison with a NaNO2 standard curve. Results are means ± 1 s.e.m. for triplicate cultures.
In vitro shedding of TNF-R75 from different cell subsets
Spleens from LCMV-infected and uninfected mice were removed, and single-cell suspensions prepared as described earlier. Cells (5 × 105) were allowed to adhere to plastic wells for 2 h, the wells were washed twice and adherent cells were incubated for 6 h at 37°C. Other splenic cells were depleted of adherent cells by incubation in plastic bottles for 2 × 1 h, and then 5 × 105 cells were incubated for 6 h at 37°C. Finally some splenic cells were sorted in CD8/non-CD8 by flow cytometry using a Becton Dickinson (San Jose, CA) FACStar Plus [17,32]. Splenic cells were stained with PE anti-CD8a and FITC anti-Mac-1, and Mac-1− cells were sorted into CD8+ and CD8− cells. Postsort analyses were performed to determine sort purity; the purity of all populations was > 90%. Cells (3.5 × 105) were incubated for 8 h at 37°C. Supernatants were harvested and assayed for soluble TNF-R75.
Quantification of cytokine and cytokine receptor levels in serum and culture supernatants
All cytokine and cytokine receptor concentrations were assayed using sandwich ELISAs. The following ELISA kits were used in this study: TNF-α (Endogen, Cambridge, MA), TNF-RI (p55), and TNF-RII (p75) (Genzyme, Cambridge, MA). Assays were run according to the manufacturer's instructions. Duplicate samples were analysed for each serum, and for cell cultures supernatants harvested from duplicate or triplicate wells were assayed individually.
Endotoxic shock
Mice were challenged with 10 μg of LPS intraperitioneally, and mortality was recorded after 24 h and 48 h.
RESULTS
Virus-induced changes in sTNF-R55 and sTNF-R75 in serum
To study the influence of viral infection on sTNF-R levels in serum, mice were infected with either of two viruses of markedly different biology. LCMV is a natural mouse pathogen that replicates extensively in all the major organs inducing a potent host response dominated by activation of CD8+ T cells [17,23,33]. In contrast, VSV is a neurotropic virus that induces a much more limited immune response [30]. However, a substantial innate immune response is observed, and around day 6 post-infection (p.i.) a cytotoxic T cell response can be detected [26–28]. Groups of mice were infected intravenously with either LCMV or VSV, and on the indicated days, mice were bled and serum was isolated and stored at −70°C until analysis. The serum concentration of sTNF-R55 in uninfected mice was low (0.31 ng/ml, median of five mice) compared with the level of sTNF-R75 (2.48 ng/ml, median of five mice), and we found no significant increase of sTNF-R55 in serum of neither LCMV-infected nor VSV-infected mice (data not shown). However, as seen in Fig. 1a, the serum concentration of sTNF-R75 in LCMV-infected mice was slightly elevated on day 3 p.i., peaked around day 8 p.i. and had almost returned to background about 2 weeks after virus inoculation. Additional analysis confirmed that sTNF-R75 levels in serum of LCMV-infected mice peaked on day 8 p.i. with intermediate levels on days 5 and 11 p.i. (data not shown). In VSV-infected mice a different pattern was observed: serum levels of sTNF-R75 peaked on day 1 p.i., but then rapidly declined and were only moderately elevated on day 6 p.i. (Fig. 1c). These patterns of TNF-R shedding suggest a role of both innate and specific immunity in regulating production of sTNF-R75 induced as a result of viral infection.
Fig. 1.
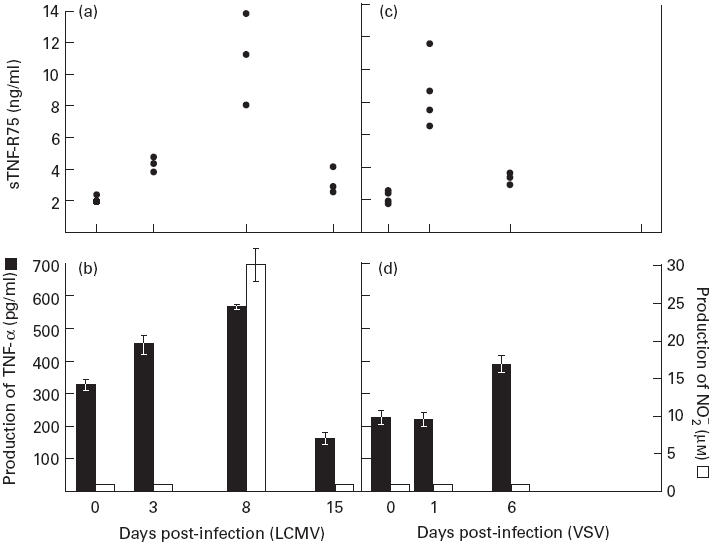
Kinetics of changes in the level of sTNF-R75 in serum of virus-infected mice (a,c), compared with in vitro lipopolysaccharide (LPS)-induced production of TNF-α and nitric oxide (NO) from splenic adherent cells, isolated from the same mice (b,d). Points represent individual mice, and bars are means ± s.e.m. of groups. One of two identical experiments is shown.
Since the above kinetics are reminiscent of the pattern previously observed with regard to macrophage activation in LCMV-infected mice [19,24], we compared the shedding of TNF-R75 with in vitro production of TNF-α and NO by splenic adherent cells. In LCMV-infected mice, we found a good correlation between shedding of TNF-R75 and activation of macrophages (Fig. 1a,b). Thus, production of TNF-α was slightly elevated 3 days after infection, peaked on day 8 p.i. and was again at base level 15 days p.i. (the difference between day 0 and day 15 values observed in this experiment was not reproducible upon repetition). Production of NO was measurable only on day 8 p.i., in keeping with the known role of T cells in LCMV-induced activation of macrophages [24]. In VSV-infected mice, however, release of TNF-α was only significantly increased on day 6 p.i., and there was no measurable NO production either on day 1 or day 6 p.i. (Fig. 1d). Consequently, the burst of sTNF-R75 on day 1 p.i. must have been caused by an innate host response not including substantial activation of splenic macrophages. The less pronounced T cell activation in VSV-infected mice probably explains both the lower levels of sTNF-R75 in serum on day 6 p.i. as well as marginal production of TNF-α and NO by splenic adherent cells in vitro.
Role of T cells in the LCMV-induced increase in sTNF-R75
The fact that peak levels of sTNF-R75 in serum of LCMV-infected mice were observed on day 8 p.i., coinciding with maximal T cell-mediated immunity, suggested a critical role of T cells in regulating this response. To verify that, matched groups of T cell-deficient nu/nu mice and wild-type mice were infected, and serum was assayed for sTNF-R75 levels on day 8 p.i. (Fig. 2a). We found a significant difference in sTNF-R75 concentration in serum between the two strains, with levels of sTNF-R75 in infected nu/nu mice being approx. three-to-four-fold lower than in infected wild-type mice, almost equalling levels in uninfected nu/nu and wild-type controls. The level of TNF-α and NO produced in vitro by splenic adherent cells from the same mice correlated well with sTNF-R75 in serum (Fig. 2b). The fact that both TNF-α and NO were produced at basal levels in LCMV-infected nu/nu mice confirms the pivotal role of T cells in LCMV-induced macrophage activation as found earlier [19,34,35].
Fig. 2.
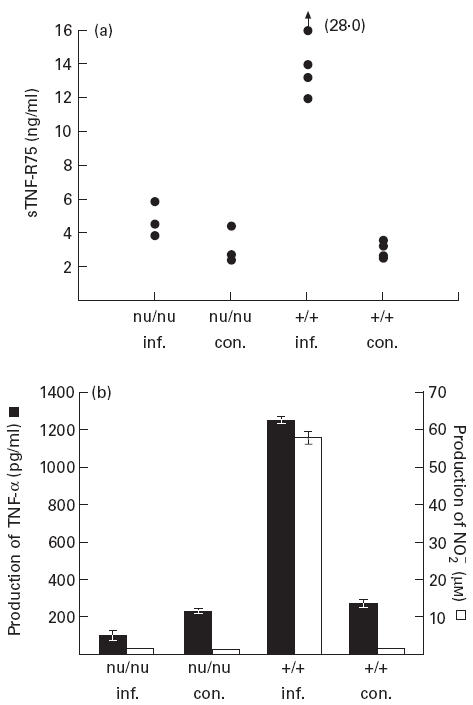
Lymphocytic choriomeningitis virus (LCMV)-induced increase in sTNF-R75 levels in serum (a), as well as in production of TNF-α and nitric oxide (NO) by splenic adherent cells in vitro (b), is T cell-dependent. LCMV-infected (inf.) T cell-deficient (nu/nu) mice and wild-type (+/+) mice were killed 8 days post-infection together with uninfected control mice (con.) from both groups. Points represent individual mice, and bars are means ± s.e.m. of groups.
Role of IFN-γ and CD40 ligand in the LCMV-induced increase in sTNF-R75
In an attempt to define more precisely the mechanism by which T cells influence the level of sTNF-R75 in serum and to evaluate the role of activated macrophages in this respect, we investigated mice deficient in CD40 ligand (CD40L −/−) and IFN-γ (IFN-γ −/−), respectively. Both molecules have previously been shown to play an important role in macrophage activation during infection [26,36–39]. CD40L −/− mice and IFN-γ −/− mice were infected in parallel with wild-type controls, and 8 days later all the mice were bled, and the level of sTNF-R75 in serum was quantified. Surprisingly, essentially similar increases in serum levels of sTNF-R75 were observed in infected mutants and infected wild-type mice (Fig. 3a), indicating a CD40L-, as well as an IFN-γ-independent mechanism of increased shedding of TNF-R75 in LCMV-infected mice. We also found that the level of TNF-α and NO produced in vitro by splenic adherent cells from IFN-γ-deficient mice did not correlate with levels of sTNF-R75, as there was no production of either TNF-α or NO above the basal level (Fig. 3b). This points to suboptimal activation of macrophages in these mice and suggests that sTNF-R75 is either the product of another cell subset or that macrophages, not fully activated, are able to shed TNF-R75. TNF-α and NO levels from CD40L −/− mice correlated with sTNF-R75 levels (Fig. 3b). Thus CD40L did not seem to be required for LCMV-induced macrophage activation: although the production of NO by splenic adherent cells tended to be slightly lower in CD40L −/− mice, the difference was not statistically significant (Mann–Whitney rank test, P > 0.05).
Fig. 3.
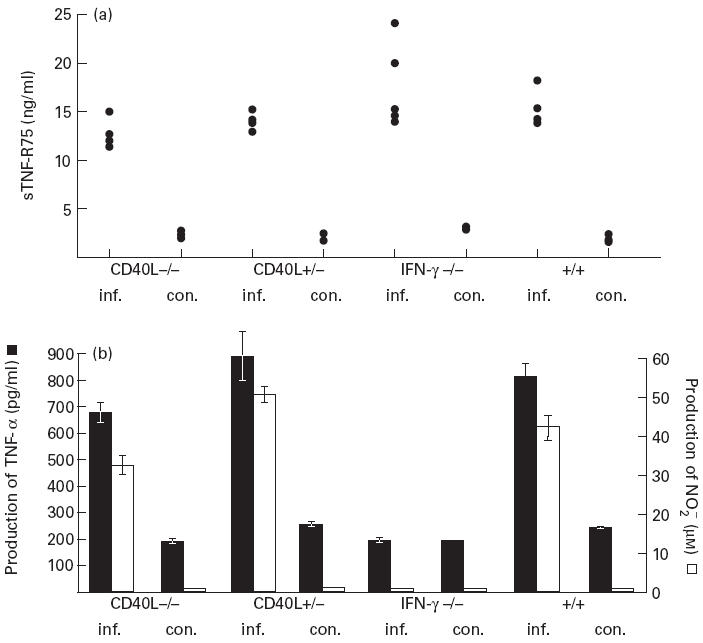
Role of CD40 ligand and IFN-γ in lymphocytic choriomeningitis virus (LCMV)-induced increase in sTNF-R75 levels in serum (a), as well as in production of TNF-α and nitric oxide (NO) by splenic adherent cells in vitro (b). LCMV-infected (inf.) CD40 ligand-deficient (CD40L−/−) mice, CD40L+/− female mice, IFN-γ-deficient (IFN-γ −/−) mice and wild-type (+/+) mice were killed and tested 8 days post-infection, together with uninfected control mice (con.) from both groups. Points represent individual mice, and bars are means ± s.e.m. of groups.
In vitro shedding of TNF-R75 from different spleen cell subsets
The absence of a simple correlation between increased shedding of TNF-R75 and the state of macrophage activation in the spleen of LCMV-infected mice (which is a major site of LCMV replication) raised the question whether other cell subsets, e.g. T cells [40], were contributing to the observed increase.
Separating spleen cells into adherent and non-adherent cells, we found that both cell populations from LCMV-infected mice released increased amounts of sTNF-R75 into the medium, compared with similarly separated cells from uninfected controls (Fig. 4a). Considering the ratio of adherent to non-adherent cells in mixed spleen cell populations (approx. 1:3–4), adherent cells appear to have a higher production-rate of sTNF-R75, probably due to a higher cell surface expression. Of non-macrophage (Mac-1−) spleen cells, CD8+ cells from infected animals were the only cell subset giving rise to an increase in sTNF-R75 in culture supernatants (Fig. 4b).
Fig. 4.
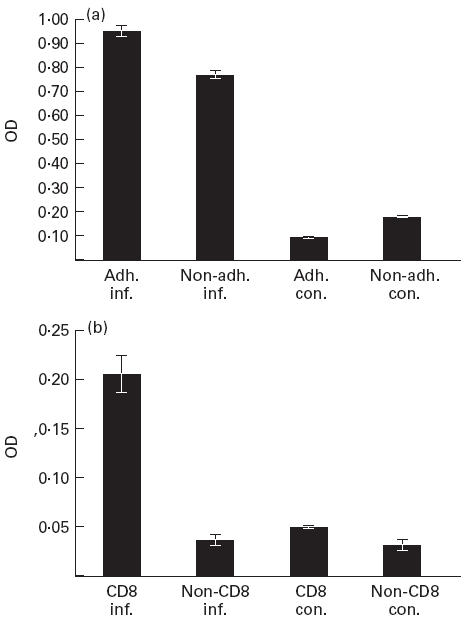
In vitro production of sTNF-R75 in culture supernatants from adherent (adh.) and non-adherent (non-adh.) splenic cells (a), as well as from Mac-1+ depleted CD8+ and non-CD8+ cell populations (b). Cells (5 × 105/well) were incubated for 6 h (a), and 3.2 × 105 cells/well were incubated for 8 h (b). sTNF-R75 concentrations are given as optical density (OD) values; bars represent means ± s.e.m. of groups. Mice were either infected with 103 LD50 of lymphocytic choriomeningitis virus (LCMV) intravenously 8 days previously, or left untreated for control.
In order to test specifically whether IFN-γ is a critical regulator for the shedding of sTNF-R75 from monocytes/macrophages, adherent cells from LCMV-infected wild-type mice and IFN-γ −/− mice were directly compared with regard to shedding of this receptor into cell culture supernatants. In two independent experiments we found that no increased release of sTNF-R75 was detected in adherent cell cultures from infected IFN-γ −/− mice relative to uninfected animals, whereas this was observed consistently with parallel cultures from infected wild-type mice (data not shown).
LPS-induced production of sTNF-R75
Soluble TNF-R75 is hypothesized to function as an antagonist to TNF-α [41–43]. It is well known that i.p. injection of LPS—the main initiator of septic shock—results in hyperproduction of especially TNF-α by activated macrophages. It has also been found that systemic infection with LCMV sensitizes mice to LPS [19]. Therefore, we investigated the serum levels of sTNF-R75 and TNF-α in mice sensitized to LPS. Mice were infected with LCMV intravenously, and 8 days post-infection they received a dose of 10 μg LPS intraperitoneally. This combination is known to cause a lethal septic shock in virus-infected immunocompetent mice resulting in death 6–24 h after LPS injection [19]. For comparison, a group of uninfected mice received the same dose of LPS and unchallenged infected and uninfected mice were used as controls. Blood samples were taken 1.5 h after LPS challenge and frozen at −70°C until analysed for sTNF-R75 and TNF-α. Mortality was recorded 24 h and 48 h after LPS challenge.
Hypersensitivity to LPS in LCMV-infected mice correlated with hyperproduction of sTNF-R75 as well as TNF-α in serum (Fig. 5). Serum levels of sTNF-R75 were approx. 10-fold higher in infected mice challenged with LPS compared with infected unchallenged mice. The same difference was observed between LPS-challenged and unchallenged uninfected mice. The level of TNF-α in serum was only detectable in LPS-challenged mice, and was markedly increased (> 20-fold) in mice infected with LCMV 8 days earlier compared with uninfected mice. The ratio between TNF-α and TNF-R75 was calculated for the two groups of LPS-challenged mice, and the result was evaluated in relation to the observed mortality. Only mice infected with LCMV before LPS challenge died, and the ratio between TNF-α and sTNF-R75 was much higher in virus-infected (LPS-sensitized) mice compared with uninfected mice. This observation is in keeping with the hypothesis of sTNF-Rs functioning as antagonists to TNF-α.
Fig. 5.
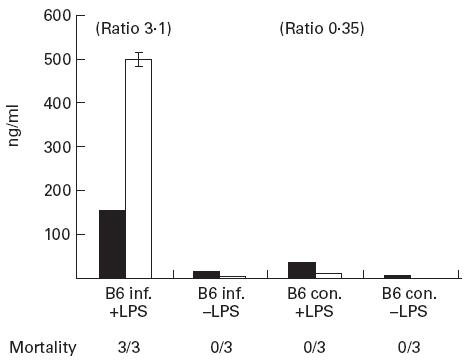
Lipopolysaccharide (LPS)-induced production of sTNF-R75 and TNF-α in serum of lymphocytic choriomeningitis virus (LCMV)-infected (inf.) and uninfected control (con.) mice. Bars represent means ± s.e.m. of the groups. Ratios between TNF-α (□) and sTNF-R75 (▪) are given above the bars for LPS-stimulated groups, and mortality is stated for each group.
Shedding of TNF-R75 by innate immune mechanisms: role of IFN-α/β
The observed increase in sTNF-R75 in serum already 1 day after infection with VSV indicated that sTNF-R75 would also serve as a marker of the innate host response. For this reason we tested whether the IFN-inducer Poly I:C would have a similar effect. Similar to what was observed in VSV-infected mice, pretreatment with Poly I:C resulted in an approx. three-fold increase in level of sTNF-R75 in serum (Fig. 6) This suggested to us that IFN-α/β might be a critical mediator in this early response, and therefore IFN-α/β receptor knockout mice were pretreated with Poly I:C, and the increase in sTNF-R75 level was evaluated. We found virtually no increased shedding of sTNF-R75 in pretreated IFN-α/βR−/− mice, compared with matched wild-type animals (129Sv). Essentially similar results were obtained in VSV-infected IFN-α/βR−/− mice. In contrast, the early increase in sTNF-R75 levels observed in serum 1 day post-VSV infection was clearly T cell-independent (Fig. 6), as evidenced by the finding that levels in infected nu/nu mice were comparable with levels in infected wild-type mice.
Fig. 6.
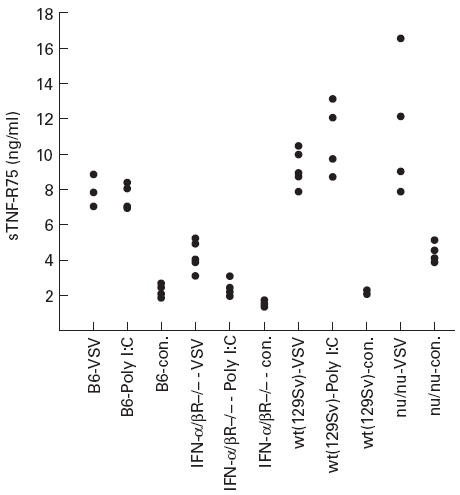
Shedding of TNF-R75 in the non-adaptive phase; role of IFN-α/β. Serum levels of sTNF-R75 were determined in serum taken 1 day post an i.v. vesicular stomatitis virus (VSV) infection or 1 day post an i.p. polyinosinic-polycytidylic acid (Poly I:C) injection in either C57Bl/6 mice (B6), IFN-α/β receptor-deficient mice (IFN-α/βR−/−), 129Sv wild-type mice (wt(129Sv)) or T cell-deficient nude mice (nu/nu). Uninfected mice from each strain served as controls (con.). Points represent individual mice.
DISCUSSION
Using two very different viral infections, we have clearly demonstrated that an increase in sTNF-R75 level in serum may reflect at least two different types of anti-viral host reactivity. Thus an increase in sTNF-R75 level may be found both early after virus inoculation, reflecting perturbation of innate resistance mechanism, and later coinciding with the appearance of a specific, cell-mediated immune response. In contrast, the level of sTNF-R55 was of no predictive value in either case, since no increased shedding of this soluble receptor was observed at any time point in virus-infected mice. Analysis of infected mice at the time point of maximal shedding of sTNF-R75 due to LCMV infection revealed that this increased shedding was dependent on the presence of T cells (Fig. 2a), which is in keeping with the commonly held notion that a rise in serum level of sTNF-R75 may be used as a marker for a cell-mediated immune response [12]. However, contrary to our expectations, we found that increased serum levels were induced independently of expression of both CD40 ligand and IFN-γ (Fig. 3a). The last observation is at variance with previous findings in human studies in which sTNF-R levels in serum generally correlate strongly with IFN-γ [12,44], and sTNF-R75 is used as an adjunct for quantification of Th1-type immune responses [12]. Comparing sTNF-R75 levels in serum with the degree of macrophage activation evaluated in vitro, we have made three pertinent observations. First, LCMV-mediated activation of splenic macrophages is critically dependent upon IFN-γ, as production of TNF-α and NO by splenic adherent cells from infected IFN-γ −/− mice is of similar magnitude to that produced by cells from uninfected IFN-γ −/− and wild-type control mice (Fig. 3b). Second, CD40 ligand does not seem to play any role in LCMV-induced macrophage activation. This is in contrast to what has previously been found when analysing the murine Leishmania infection [45], and could suggests that alternative mediators may be active during a systemic viral infection, making signalling through CD40/CD40L interaction redundant. Third, contrary to our working hypothesis, shedding of sTNF-R75 into serum does not correlate directly with macrophage activation. This is evidenced by two findings. First, sTNF-R75 is found in significantly increased amounts in serum of mice infected with VSV 1 day earlier, at a time point when no macrophage activation could be demonstrated in vitro. This finding should, however, be interpreted with care. Thus, in vitro we have only analysed the activation state of splenic macrophages, and other cellular compartments (e.g. Kupffer cells in the liver) may be more important, especially in the case of the VSV infection, in which virus does not replicate extensively in wild-type mice. Second, levels of sTNF-R75 in serum of LCMV-infected IFN-γ −/− mice are comparable to levels found in infected wild-types. In this case, however, sTNF-R75 shedding from splenic adherent cells does seem to correlate with macrophage activation as evaluated in vitro, which may suggest that the regulation of serum levels is complex and the net result of shedding from several different cellular sources. Consistent with this interpretation our study of TNF-R75 shedding in vitro revealed that activated CD8+ T cells from LCMV-infected mice may constitute an important cellular source to the pool of sTNF-R75, although on a cell-for-cell basis splenic adherent cells seem to have a higher production rate of this soluble receptor. Yet, in view of the fact that activated CD8+ T cells make up about one third or more of the spleen cells in LCMV-infected mice around day 8 p.i. [17], it is tempting to suggest that CD8+ T cells are major contributors to the increased serum level of sTNF-R75 observed during the specific immune response to LCMV infection. In keeping with this suggestion, little increase in sTNF-R75 level is found in serum of mice infected with VSV 6 days earlier, and in these mice a much less substantial increase in numbers of activated CD8+ T cells is seen (unpublished observations).
Studies of TNF-R75 shedding early after LCMV or VSV infection disclosed that also innate immune mechanisms may cause an increase in the level of sTNF-R75 in serum during viral infection. The major mediator in this context appears to be IFN-α/β (Fig. 6). First, similar results may be obtained by injection of the IFN-inducer Poly I:C, and, more importantly, little or no increase is seen in IFN-α/βR−/− mice using either Poly I:C or VSV. Notably, when mice are injected with viruses, Poly I:C or LPS, a rapid CD8+CD44high proliferative response is induced; this is not seen in IFN-α/βR−/− mice [46,47], indicating a role for IFN-α/β in activation of primed T cells. However, experiments with VSV-infected, T cell-deficient mice revealed that serum levels of sTNF-R75 in these mice 1 day after infection were comparable to those in similarly infected wild-type mice (Fig. 6). Thus, the increase in sTNF-R75 early after virus infection, although mediated via IFN-α/β, does not require T cells. Therefore, it appears that in addition to the different phases in shedding of TNF-R75 during virus infection, T cell-dependent as well as T cell-independent mechanisms may be operative.
From recent studies on susceptibility to septic shock in virus-infected mice, it is known that mice are sensitized to LPS with maximal susceptibility 8 days after an LCMV infection and 1 day after a VSV infection ([19], unpublished data). Since peak levels of sTNF-R75 are observed on the very same days following the two infections, respectively, sTNF-R75 could be used clinically as a marker for the altered state underlying an increased susceptibility to septic shock. The difference in ratios between TNF-α and sTNF-R75 in LPS-sensitized and non-sensitized mice compared with mortality, support the hypothesis that sTNF-Rs may function as an antagonist to TNF-α, reducing its proinflammatory and toxic activity.
In conclusion, we have shown that serum levels of sTNF-Rs are markers of the innate as well as the specific cell-mediated host response to a viral infection. Increased release of sTNF-R75 into serum during the cell-mediated immune response is mediated via T cells, but is independent of CD40 ligand and IFN-γ. During the innate immune response to viral infection, T cells are of no importance, whereas IFN-α/β seems to be the major mediator. The level of sTNF-Rs in serum may therefore be a sensitive marker of host reactivity during viral infection, but it is not correlated to a single immunological parameter, since several different mechanisms may cause an increase. Whether similar mechanisms are operating in humans needs to be explored, but conclusions about diseases based on sTNF-R levels alone ought to be interpreted with caution.
Acknowledgments
A.N. is the recipient of a PhD scholarship from the Faculty of Health Sciences, University of Copenhagen. This work was supported in part by the Danish Medical Reasearch Council, the Biotechnology Centre for Cellular Communication, the Novo Nordisk Foundation, the Association for Rheumatoid Arthritis, and the Foundation for Advancement of Medical Science.
REFERENCES
- 1.Rothe J, Lesslauer W, Lotscher H, et al. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 2.Pfeffer K, Matsuyama T, Kundig TM, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–67. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 3.Peschon JJ, Torrance DS, Stocking KL, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–52. [PubMed] [Google Scholar]
- 4.Erickson SL, de Sauvage FJ, Kikly K, et al. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 1994;372:560–3. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- 5.Tsujimoto M, Yip YK, Vilcek J. Interferon-gamma enhances expression of cellular receptors for tumor necrosis factor. J Immunol. 1986;136:2441–4. [PubMed] [Google Scholar]
- 6.Ruggiero V, Tavernier J, Fiers W, et al. Induction of the synthesis of tumor necrosis factor receptors by interferon-gamma. J Immunol. 1986;136:2445–50. [PubMed] [Google Scholar]
- 7.Aderka D, Englemann H, Hornik V, et al. Increased serum levels of soluble receptors for tumor necrosis factor in cancer patients. Cancer Res. 1991;51:5602–7. [PubMed] [Google Scholar]
- 8.Aukrust P, Liabakk NB, Muller F, et al. Serum levels of tumor necrosis factor-alpha (TNF alpha) and soluble TNF receptors in human immunodeficiency virus type 1 infection—correlations to clinical, immunologic, and virologic parameters. J Inf Dis. 1994;169:420–4. doi: 10.1093/infdis/169.2.420. [DOI] [PubMed] [Google Scholar]
- 9.Zangerle R, Gallati H, Sarcletti M, et al. Tumor necrosis factor alpha and soluble tumor necrosis factor receptors in individuals with human immunodeficiency virus infection. Immunol Letters. 1994;41:229–34. doi: 10.1016/0165-2478(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 10.Kalinkovich A, Engelmann H, Harpaz N, et al. Elevated serum levels of soluble tumour necrosis factor receptors (sTNF-R) in patients with HIV infection. Clin Exp Immunol. 1992;89:351–5. doi: 10.1111/j.1365-2249.1992.tb06961.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Poll T, Jansen J, van Leenen D, et al. Release of soluble receptors for tumor necrosis factor in clinical sepsis and experimental endotoxemia. J Inf Dis. 1993;168:955–60. doi: 10.1093/infdis/168.4.955. [DOI] [PubMed] [Google Scholar]
- 12.Diez-Ruiz A, Tilz GP, Zangerle R, et al. Soluble receptors for tumour necrosis factor in clinical laboratory diagnosis. Eur J Haematol. 1995;54:1–8. doi: 10.1111/j.1600-0609.1995.tb01618.x. [DOI] [PubMed] [Google Scholar]
- 13.Miyawaki T, Kasahara Y, Kanegane H, et al. Expression of CD45RO (UCHL1) by CD4+ and CD8+ T cells as a sign of in vivo activation in infectious mononucleosis. Clin Exp Immunol. 1991;83:447–51. doi: 10.1111/j.1365-2249.1991.tb05659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward BJ, Johnson RT, Vaisberg A, et al. Spontaneous proliferation of peripheral mononuclear cells in natural measles virus infection: identification of dividing cells and correlation with mitogen responsiveness. Clin Immunol Immunopathol. 1990;55:315–26. doi: 10.1016/0090-1229(90)90107-2. [DOI] [PubMed] [Google Scholar]
- 15.Griffin DE, Ward BJ, Jauregui E, et al. Immune activation during measles: interferon-gamma and neopterin in plasma and cerebrospinal fluid in complicated and uncomplicated disease. J Inf Dis. 1990;161:449–53. doi: 10.1093/infdis/161.3.449. [DOI] [PubMed] [Google Scholar]
- 16.Carding SR, Allan W, McMickle A, et al. Activation of cytokine genes in T cells during primary and secondary murine influenza pneumonia. J Exp Med. 1993;177:475–82. doi: 10.1084/jem.177.2.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersson EC, Christensen JP, Marker O, et al. Changes in cell adhesion molecule expression on T cells associated with systemic virus infection. J Immunol. 1994;152:1237–45. [PubMed] [Google Scholar]
- 18.Colle JH, Saron MF, Shidani B, et al. High frequency of T lymphocytes committed to interferon-gamma transcription upon polyclonal activation in spleen from lymphocytic choriomeningitis virus-infected mice. Int Immunol. 1993;5:435–41. doi: 10.1093/intimm/5.5.435. [DOI] [PubMed] [Google Scholar]
- 19.Nansen A, Christensen JP, Marker O, et al. Sensitization to lipopolysaccharide in mice with asymptomatic viral infection: role of T cell-dependent production of interferon-gamma. J Inf Dis. 1997;176:151–7. doi: 10.1086/514017. [DOI] [PubMed] [Google Scholar]
- 20.Christensen JP, Ropke C, Thomsen AR. Virus-induced polyclonal T cell activation is followed by apoptosis: partitioning of CD8+ T cells based on alpha 4 integrin expression. Int Immunol. 1996;8:707–15. doi: 10.1093/intimm/8.5.707. [DOI] [PubMed] [Google Scholar]
- 21.Akbar AN, Borthwick N, Salmon M, et al. The significance of low bcl-2 expression by CD45RO T cells in normal individuals and patients with acute viral infections. The role of apoptosis in T cell memory. J Exp Med. 1993;178:427–38. doi: 10.1084/jem.178.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasaian MT, Biron CA. The activation of IL-2 transcription in L3T4+ and Lyt-2+ lymphocytes during virus infection in vivo. J Immunol. 1989;142:1287–92. [PubMed] [Google Scholar]
- 23.Murali-Krishna K, Altman JD, Suresh M, et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–87. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 24.Buchmeier MJ, Welsh RM, Dutko FJ, et al. The virology and immunobiology of lymphocytic choriomeningitis virus infection. [Review] [180 refs] Adv Immunol. 1980;30:275–331. doi: 10.1016/s0065-2776(08)60197-2. [DOI] [PubMed] [Google Scholar]
- 25.Thomsen AR, Pfau CJ. Influence of host genes on the outcome of murine lymphocytic choriomeningitis virus infection. In: Salvato MS, editor. The arenaviridae. New York: Plenum Press; 1993. pp. 199–224. [Google Scholar]
- 26.Muller U, Steinhoff U, Reis LF, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 27.Zinkernagel RM, Althage A, Holland J. Target antigens for H-2-restricted vesicular stomatitis virus-specific cytotoxic T cells. J Immunol. 1978;121:744–8. [PubMed] [Google Scholar]
- 28.Schattner A, Meshorer A, Wallach D. Involvement of interferon in virus-induced lymphopenia. Cell Immunol. 1983;79:11–25. doi: 10.1016/0008-8749(83)90046-1. [DOI] [PubMed] [Google Scholar]
- 29.Marker O, Volkert M. Studies on cell-mediated immunity to lymphocytic choriomeningitis virus in mice. J Exp Med. 1973;137:1511–25. doi: 10.1084/jem.137.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomsen AR, Nansen A, Andersen C, et al. Cooperation of B cells and T cells is required for survival of mice infected with vesicular stomatitis virus. Int Immunol. 1997;9:1757–66. doi: 10.1093/intimm/9.11.1757. [DOI] [PubMed] [Google Scholar]
- 31.Nansen A, Christensen JP, Ropke C, et al. Role of interferon-γ in the pathogenesis of LCMV-induced meningitis: unimpaired leucocyte recruitment, but deficient macrophage activation in interferon-γ knock-out mice. J Neuroimmunol. 1998;86:202–12. doi: 10.1016/s0165-5728(98)00055-1. [DOI] [PubMed] [Google Scholar]
- 32.Christensen JP, Andersson EC, Scheynius A, et al. Alpha 4 integrin directs virus-activated CD8+ T cells to sites of infection. J Immunol. 1995;154:5293–301. [PubMed] [Google Scholar]
- 33.Andersson EC, Christensen JP, Scheynius A, et al. Lymphocytic choriomeningitis virus infection is associated with long-standing perturbation of LFA-1 expression on CD8+ T cells. Scand J Immunol. 1995;42:110–8. doi: 10.1111/j.1365-3083.1995.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 34.Butz EA, Hostager BS, Southern PJ. Macrophages in mice acutely infected with lymphocytic choriomeningitis virus are primed for nitric oxide synthesis. Microbial Pathogenesis. 1994;16:283–95. doi: 10.1006/mpat.1994.1029. [DOI] [PubMed] [Google Scholar]
- 35.Leist TP, Heuchel R, Zinkernagel RM. Increased bactericidal macrophage activity induced by immunological stimuli is dependent on interferon (IFN)-gamma. Interference of anti-IFN-gamma but not anti-IFN-alpha/beta with modulation of macrophage activity caused by lymphocytic choriomeningitis virus infection or systemic graft-vs.-host reactions. Eur J Immunol. 1988;18:1295–8. doi: 10.1002/eji.1830180822. [DOI] [PubMed] [Google Scholar]
- 36.Leist TP, Heuchel R, Zinkernagel RM. Increased bactericidal macrophage activity induced by immunological stimuli is dependent on interferon (IFN)-gamma. Interference of anti-IFN-gamma but not anti-IFN-alpha/beta with modulation of macrophage activity caused by lymphocytic choriomeningitis virus infection or systemic graft-vs.-host reactions. Eur J Immunol. 1988;18:1295–8. doi: 10.1002/eji.1830180822. [DOI] [PubMed] [Google Scholar]
- 37.Stout RD. Macrophage activation by T cells: cognate and non-cognate signals. Curr Opin Immunol. 1993;5:398–403. doi: 10.1016/0952-7915(93)90059-2. [DOI] [PubMed] [Google Scholar]
- 38.Stout RD, Suttles J. The many roles of CD40 in cell-mediated inflammatory responses. Immunol Today. 1996;17:487–92. doi: 10.1016/0167-5699(96)10060-i. [DOI] [PubMed] [Google Scholar]
- 39.Dalton DK, Pitts-Meek S, Keshav S, et al. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739–42. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 40.Cope AP, Aderka D, Wallach D, et al. Soluble TNF receptor production by activated T lymphocytes: differential effects of acute and chronic exposure to TNF. Immunology. 1995;84:21–30. [PMC free article] [PubMed] [Google Scholar]
- 41.Girardin E, Roux-Lombard P, Grau GE, et al. Imbalance between tumour necrosis factor-alpha and soluble TNF receptor concentrations in severe meningococcaemia. The J5 Study Group. Immunology. 1992;76:20–23. [PMC free article] [PubMed] [Google Scholar]
- 42.van Deuren M, van der Ven-Jongekrijg J, Demacker PN, et al. Differential expression of proinflammatory cytokines and their inhibitors during the course of meningococcal infections. J Infect Dis. 1994;169:157–61. doi: 10.1093/infdis/169.1.157. [DOI] [PubMed] [Google Scholar]
- 43.Carpenter A, Evans TJ, Buurman WA, et al. Differences in the shedding of soluble TNF receptors between endotoxin-sensitive and endotoxin-resistant mice in response to lipopolysaccharide or live bacterial challenge. J Immunol. 1995;155:2005–12. [PubMed] [Google Scholar]
- 44.Zangerle R, Fuchs D, Sarcletti M, et al. Increased concentrations of soluble tumor necrosis factor receptor 75 but not of soluble intercellular adhesion molecule-1 are associated with the decline of CD4+ lymphocytes in HIV infection. Clin Immunol Immunopathol. 1994;72:328–34. doi: 10.1006/clin.1994.1149. [DOI] [PubMed] [Google Scholar]
- 45.Soong L, Xu JC, Grewal IS, et al. Disruption of CD40–CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity. 1996;4:263–73. doi: 10.1016/s1074-7613(00)80434-3. [DOI] [PubMed] [Google Scholar]
- 46.Tough DF, Sun S, Sprent J. T cell stimulation in vivo by lipopolysaccharide (LPS) J Exp Med. 1997;185:2089–94. doi: 10.1084/jem.185.12.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 1996;272:1947–50. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]