

Abstract
The aetiopathogenesis of chronic otitis media with effusion (OME) in children is not yet fully understood. OME is characterized by metaplasia of the epithelium and accumulation of sticky, glue-like effusion in the middle ear containing different mediators of inflammation, including activation fragments of the complement system. Here we examined whether the fluid phase complement activation is reflected in the middle ear mucosa and how the mucosa is protected against the cytolytic activity of complement. Mucosal biopsies from 18 middle ears of children with a history of chronic OME were taken. The biopsies were analysed by immunofluorescence microscopy after staining for complement fragments iC3b/C3c, C3d and C9, and regulators membrane cofactor protein (MCP; CD46), decay-accelerating factor (DAF; CD55) and protectin (CD59). There was a strong staining for iC3b/C3c, and a weaker one for C3d and C9 on the surface of the middle ear epithelial cells of OME patients but not in controls without OME. MCP was expressed on the hyperplastic three to four outer cell layers of the epithelium, while CD59 was expressed throughout the middle ear mucosa. The results suggest a strong ongoing complement activation and consequent inflammation in the middle ear cavity. Unrestricted complement damage of the epithelial lining is prevented by the strong expression of MCP and CD59.
Keywords: otitis media, complement activation, CD59, MCP, middle ear mucosa
INTRODUCTION
Chronic otitis media with effusion (OME) refers to the presence of middle ear effusion (MEE) in the tympanic cavity without signs or symptoms of an acute infection [1]. OME is the leading cause for elective surgery for children, and tympanostomy tubes are installed in > 500 000 children with chronic OME every year in the USA alone [2]. The aetiology of chronic OME is multifactorial and its precise pathogenic mechanisms are not yet fully understood. The most important aetiologic factors nowadays are considered to be subtle local and general immunological disturbances as well as an impaired tubal function at the age of the highest frequency of chronic OME [3,4]. Bacterial colonization or viruses during an acute infection may lead to the activation of immunocompetent cells in the middle ear and production of secretory IgA and IgG [5]. To what extent the immunostimulation is beneficial or harmful is not clear. While providing protection against further disease, the local immune system could also contribute to the immuno-pathology of chronic OME.
The highest risk of chronic OME at the age of 16 months and the peak incidence of acute otitis media at 12 months suggest that chronic OME is closely connected to an acute infection. Chronic OME is not, however, necessarily subsequent to inadequately treated episodes of acute otitis media, since there is often a latent period of varying length after an acute infection [6].
The complement system is a critical part of the immunological defence system against microbes. It consists of approx. 20 effector proteins and 10 control factors that work as a finely regulated cascade.
Activation of complement via either the classical pathway (e.g. by antibodies) or the alternative pathway (e.g. by microbial surfaces) leads to formation of the C3/C5 convertases C4b2a or C3bBb, respectively. In the human body, the formation and stability of these enzymes are regulated by the plasma factors C1INH, C4bp, H and by the cell membrane regulators C3b receptor (CR1; CD35), membrane cofactor protein (MCP; CD46) and decay-accelerating factor (DAF; CD55) [7]. These factors act together to prevent inappropriate complement activation, lysis and damage of host cells. In the terminal pathway the main inhibitors of the membrane attack complex (MAC) of complement are vitronectin (VN), clusterin and protectin (CD59). VN and clusterin act in the fluid phase to prevent insertion of terminal complement complexes to cell membranes. CD59 is a small (18–25 kD), glycophosphoinositol-anchored molecule that inhibits complement lysis on human cell membranes by binding to the MAC components C8 and C9 [8,9].
Activation of complement is not entirely beneficial. When activated by necrotic tissue, as occurs for example in myocardial infarction [10] or during an autoimmune attack, the complement system can extend the damage of host tissue. Engineering of complement inhibitors has become an important goal for the prevention of complement mediated-tissue damage [11].
In acute otitis media, the complement system can become activated by microbes that enter the middle ear space through the Eustachian tube. Complement components probably diffuse from the blood plasma and/or are synthesized locally, e.g. by macrophages in the tissue. In chronic OME, microbes have mostly been eliminated by different immunological defence systems, and often by antibiotics. Live bacteria can only be detected in 1/3 of middle ear effusions in chronic OME, but a strong local complement activation persists in the middle ear effusion [12].
The reasons for an ongoing complement activation in chronic OME are unclear. Potentially, complement can become activated by many different substances present in MEE, e.g. bacterial remnants, proteolytic enzymes, degradation products from host cells, immune complexes or other immune aggregates [13,14]. It has not been examined earlier whether complement activation extends from the fluid phase to the epithelium in OME. Also, it is not known whether cells in the middle ear epithelium are protected against autologous complement to the extent that they do not become damaged.
The aims of this study were to find out if complement activation in OME extends to the middle ear mucosa by looking for deposits of complement components or their breakdown products (iC3b/C3c, C3d, C9, VN) and to study how the middle ear epithelium is protected against complement attack by analysing the local expression of complement membrane regulators (MCP, DAF, CD59).
PATIENTS AND METHODS
Patients and samples
All the children (n = 14) who participated in this study, suffered from persistent OME. They were admitted to the Department of Otorhinolaryngology, University Central Hospital, Helsinki, for tympanostomy and adenoidectomy. The diagnostic criteria for inclusion of the patients to the study were chronic otitis media with effusion for at least 2 months and no symptoms of an acute infection. The study protocol was approved by the Ethical committee of the Department of Otorhinolaryngology, Helsinki University Central Hospital. The biopsies were taken with parental consent in connection with normal treatment.
Small samples of middle ear mucosa were obtained under general anaesthesia in connection with myringotomy and insertion of a ventilation tube. Samples were taken from the anterior promontorial surface with a microcup forceps through the myringotomy incision. Samples of the effusions were taken for bacterial culture. The bacterial culture was positive in 4/18 samples. The different bacteria cultured were Streptococcus pneumoniae, Staphylococcus aureus and coagulase-negative staphylococci (n = 2).
The persistence of the middle ear effusion in the patients varied from 2 to 10 months (mean 4.5 months). The total number of samples analysed was 18 since bilateral samples were obtained from four children. Ten of the children were boys and four were girls. The mean age of the patients at the time of operation was 3.6 years (range 1.6–7.5 years). The nature of the effusion was mucoid in 16/18 ears. The effusion was seromucoid in one ear and mucopurulent in another. Two samples of healthy middle ear epithelium were obtained in connection with ear operations. As it is ethically impossible to obtain samples from middle ears of healthy children, the control samples were taken in connection with other ear operations than those performed because of chronic OME. One sample was taken during a second-look operation 1 year after a congenital cholesteatoma operation from healthy middle ear epithelium of a 10-year-old boy and the other in connection with exhaeresis of the Jakobson's nerve of a 49-year-old man.
Sample processing
Biopsy specimens from the middle ear were taken into physio-logical saline and transported to the laboratory within 1 h of their removal. The samples were snap-frozen using isopentane cooled with liquid nitrogen, placed on the Tissue Tek embedding medium (Miles, Elkhart, IN) and stored at −70°C.
The frozen samples were sectioned (5 μm) in a cryostat, fixed with cold (–20°C) acetone for 5 min and rinsed three times with PBS pH 7.4. Prior to immunostaining the fixed tissue sections were treated for 5 min with 1% bovine serum albumin (BSA; Sigma Chemical Co., St Louis, MO) in PBS to prevent non-specific binding.
Immunohistochemical methods
The sections were incubated for 45 min at 22°C in a humid chamber with the following antibodies: polyclonal rabbit anti-C3c (Behringwerke AG, Marburg, Germany), goat anti-C9 (Quidel Corp., San Diego, CA), rabbit anti-vitronectin (Behringwerke); and with mouse MoAbs against C3d (4C2; a generous gift from Dr V. Koistinen in our laboratory), C9 (Quidel), MCP (CD46) (J4-48; Serotec, Oxford, UK), DAF (CD55, BRIC 216) and CD59 (BRIC 229) (both from Bio-Products Laboratory, Elstree, UK). An antibody against a neoepitope of the terminal complement complexes (Wu) was kindly obtained from Dr R. Würzner (University of Innsbruck, Austria). As controls, we used PBS, a non-specific mouse anti-idiotype MoAb AF1 (a generous gift from Dr M. Kaartinen at our Department) or heat-inactivated rabbit preimmune serum instead of the primary antibodies. After rinsing three times with PBS the sections were incubated for 30 min at 22°C with FITC-conjugated antibodies against mouse, rabbit or goat immunoglobulins (Jackson Immunoresearch Labs, West Grove, PA). After washing three times with PBS the samples were mounted with Mowiol [15]. The samples were examined using an Olympus standard microscope equipped with a filter specific for FITC fluorescence. The sections were photographed with a Nikon automatic camera on Kodak Tri-X pan or Tmax 400 ASA films.
RESULTS
Altogether, 18 middle ear biopsy specimens from 14 patients with chronic OME were analysed for the deposition of complement components (Fig. 1) or regulators (Figs 2,3 and 4). Analyses were performed on serial cryostat sections to allow comparison between individual components (Fig. 5). Immunofluorescence microscopy analysis showed that complement C3 fragments reacting with the anti-C3c antibody (C3b, iC3b and C3c) were very strongly deposited on the middle ear mucosal surface of OME patients (Fig. 1a,b). Strong granular deposits of iC3b/C3c were found in the mucus blanket on the surface of the middle ear epithelial cells. Moderate staining for iC3b/C3c was also observed on the basement membranes of the mucosal epithelium. In some sections also the capillary endothelium stained positively for iC3b/C3c. In normal middle ear mucosa (Fig. 1e) the epithelial surface did not stain positively and only a weak deposition was seen in the connective tissue. Immunostaining for the C3d fragment of C3 was found principally on the basement membranes of the mucosal epithelium and blood vessels (Fig. 5b). C3d was recognized specifically with the 4C2 MoAb that binds to C3d and C3dg, but only weakly to iC3b.
Fig. 1.
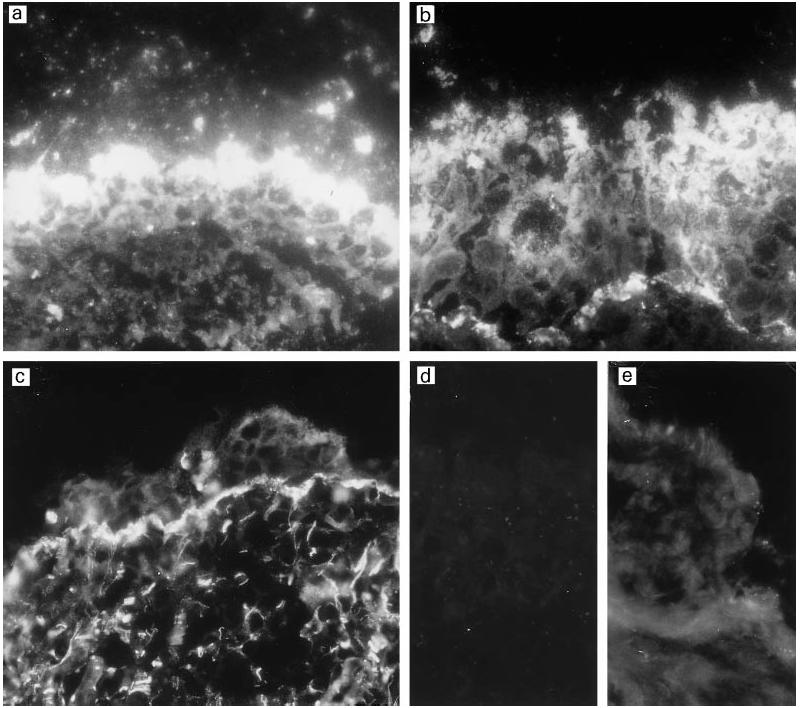
Deposition of C3 fragments on the middle ear epithelium in patients suffering from otitis media with effusion. Cryostat sections of three different middle ear biopsies were stained for iC3b/C3c with a polyclonal anti-C3c rabbit antibody (a,b) or for C3d/C3dg with the mouse MoAb 4C2 (c). iC3b/C3c is strongly deposited on the epithelial surface and on the basement membrane, whereas C3d is principally deposited on the thickened basement membrane. Some of the epithelial cells are loosely attached to the mucosal surface. In a control stained with a normal rabbit serum, no immunostaining could be detected (d). In normal middle ear epithelium stained with the C3c antibody the immunofluorescence detected was weak on the single-cell thick epithelial surface (e). (Mag. × 460.)
Fig. 2.
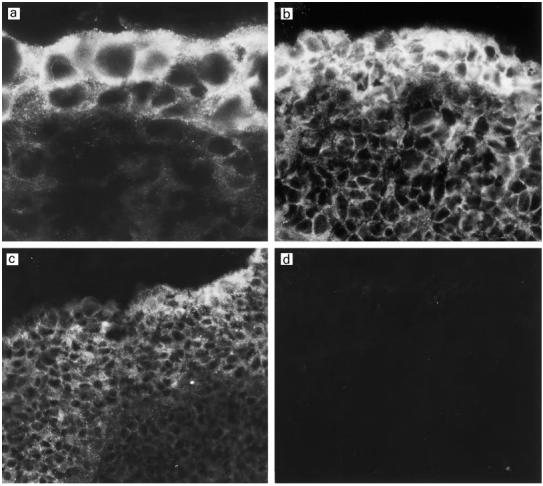
Immunostaining for the complement inhibitor membrane cofactor protein (MCP; CD46) in the middle ear mucosa of three different patients with otitis media with effusion (OME) (a–c). The expression of MCP depends on the degree of the epithelial hyperplasia which varied in thickness from two to several cell layers. MCP is expressed throughout the epithelium but the expression is strongest on the two to three outer cell layers. No staining could be detected in the controls stained with the non-specific mouse MoAb AF1 (d). (Mag.: a, × 1150; b, × 630; c–d, × 460.)
Fig. 3.
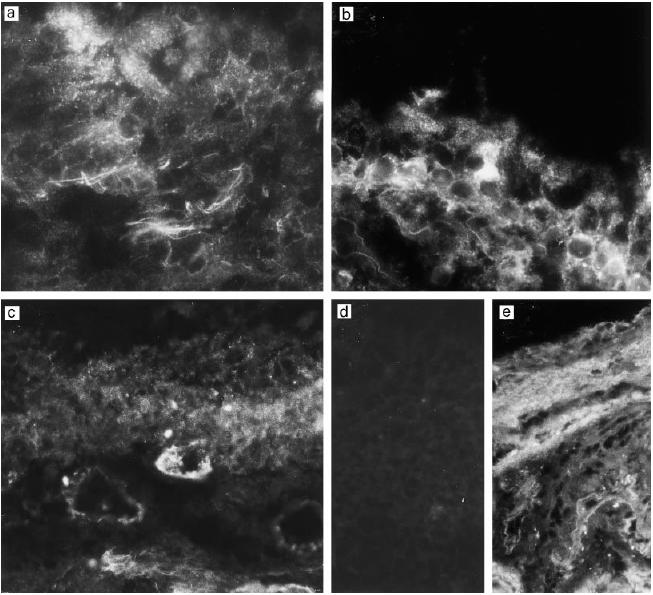
Immunostaining of the middle ear epithelium of otitis media with effusion (OME) patients for decay-accelerating factor (DAF, CD55). Samples from three different patients (a–c) show expression of DAF in connective tissue filaments, weakly in the epithelium (a,b) and occasionally in the capillary endothelia (c). No staining is seen in a control stained with the non-specific MoAb AF1 (d). In normal middle ear epithelium the staining pattern for DAF (e) is similar to that in OME patients, except that very little staining is seen in the epithelium. (Mag. × 460.)
Fig. 4.
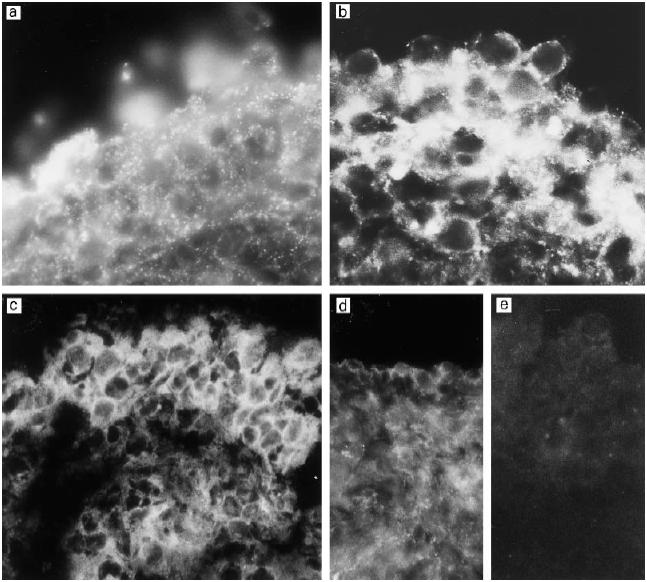
Immunostaining of middle ear biopsy specimens for the membrane attack complex inhibitor protectin (CD59). Very strong expression can be seen in samples from three different patients with otitis media with effusion (OME) (a–c). Note the granular pattern of staining (a,b). In OME patients some of the CD59 appears to be peeled off from the outermost cells of the middle ear epithelium (b). The surface of the normal middle ear epithelium also stains positively for CD59 (d). No immunostaining could be detected when the biopsies were stained with a non-specific antibody (e). (Mag.: a,b, × 690; c, × 630; d,e, × 460.)
Fig. 5.
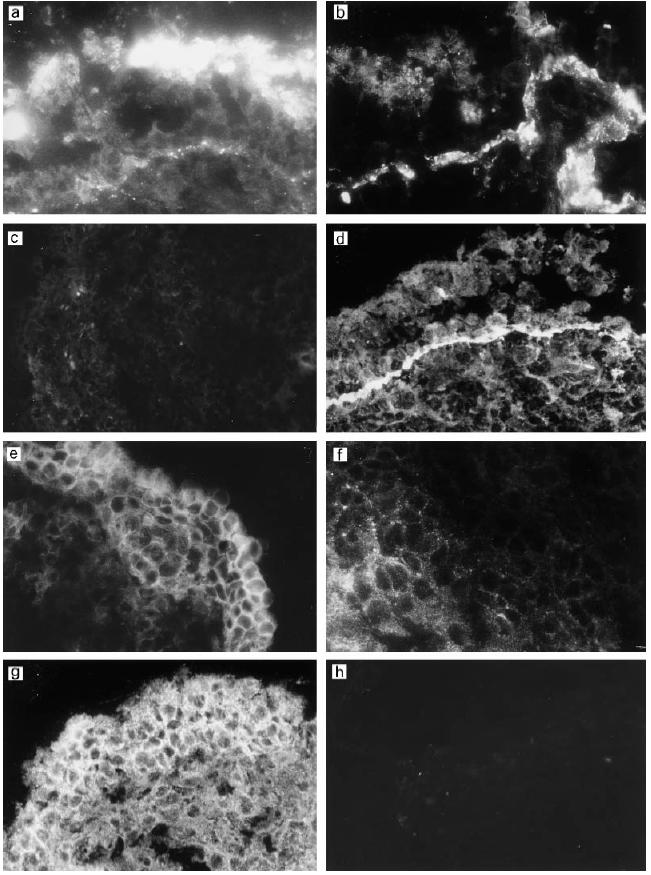
(See next page) Comparative analysis of deposition of C components and expression of C regulators in a single middle ear biopsy sample from a girl aged 2 years and 7 months. The patient had suffered from chronic otitis media with effusion (OME) for 5 months. Immunostaining for the complement components iC3b/C3c (a), C3d (b), C9 (c) and vitronectin (VN) (d) showed deposition of iC3b/C3c on the epithelial cell surface and C3d and VN on the epithelial basement membrane. Note the negative staining for C9 on the epithelium and connective tissue. The membrane regulator membrane cofactor protein (MCP) (e) was seen in the hyperplastic epithelium. The staining for decay-accelerating factor (DAF) (f) was weak but got stronger deeper in the connective tissue. CD59 (g) was strongly expressed on the epithelial cells. (Mag.: a,b,g,h, × 460; c, × 150; d, × 210; e, × 300; f, × 630.)
Analysis with both monoclonal and polyclonal anti-C9 antibodies showed only a weak or negative staining in most of the samples (Fig. 5c). When present, weak positive staining for C9 was seen on the outer surface of the middle ear epithelial cells and occasionally in the connective tissue. A similar staining pattern was seen with a terminal complement complex neoepitope-specific antibody (Wu) (not shown). The plasma inhibitor of the terminal complement cascade VN appeared positive in the basement membranes (Fig. 5d). VN could not be detected on the vascular endothelial cells, but some staining was seen in the connective tissue matrix.
In light of the strong deposition of C3 on the surface of the OME epithelium and weak staining for C9, it was of interest to analyse the expression of complement membrane regulators. A summary of the general pattern of the immunofluorescence staining of the middle ear mucosa in chronic OME is presented in Table 1. MCP (CD46) was expressed on the hyperplastic outer cell layers of the middle ear mucosa (Fig. 2). Staining on vascular endothelial cells was considerably weaker than on epithelial cells. Deeper in the connective tissue the expression of MCP was very weak.
Table 1.
Results on immunofluorescence microscopy analysis of complement components and regulators in the middle ear mucosa of patients with otitis media with effusion (OME)

*General pattern of staining intensity is shown:+++ strong staining;++ moderate;+ weak;± weak and variable;−, negative. Staining intensities should be taken to indicate differences between different tissue structures and not between individual complement components.
†Staining in connective tissue filaments.
DAF (CD55) showed a moderate or weak expression on the epithelium of the middle ear mucosa (Fig. 3). The staining pattern of healthy middle ear epithelium was similar to the OME epi-thelium, except that the epithelium in healthy middle ear is maximally only a few cell layers thick. In OME samples the outermost surface of the epithelium appeared negative for DAF and the expression got stronger towards the basal cell layers. The endothelia of capillaries stained weakly positive for DAF. In some sections, DAF appeared in connective tissue filaments. Compared with the other regulators, the distribution pattern of DAF was somewhat different. Relative to the staining in the connective tissue and endothelia the expression of DAF was weaker on the outermost surface of epithelium.
The main inhibitor of the complement membrane attack complex CD59 was found to be strongly expressed both on the surface of the epithelium and in the interstitial tissue in all samples (Fig. 4). Immunostaining of healthy middle ear epithelium showed expression of CD59 as well. CD59 was clearly present on the cell membranes in the middle ear epithelium in OME patients, but in a granular fashion. It was also found on the mucus blanket over the epithelial cells where it appeared in small granules. The vascular endothelial cells stained positive for CD59. The expression of CD59 on the outermost surface of the epithelium of chronic OME patients was weaker than in the deeper epithelial cell layers. In normal middle ear epithelium CD59 staining was seen in the single epithelial cell layer (Fig. 4d). The overall impression was that the expression of CD59 was stronger in the epithelium of OME patients than in healthy controls.
DISCUSSION
The main observation of the present study was an intense deposition of complement C3 activation fragments on the epithelium of the middle ear mucosa in children with chronic OME. Activation of complement could thus be an important factor in the pathogenesis of OME.
Strong deposition of C3 activation products on the outer surface of the middle ear epithelium indicates local complement activation in the middle ear mucosa of OME patients. This is the first study to show that complement activation occurs in the middle ear epithelium. A strong staining for C3 was seen also in small granules or vesicles in the mucous blanket on the epithelium. This may indicate that complement activation induces the epithelial cells to release small vesicles into the MEE. This finding is compatible with the previously observed extensive C3 activation in the MEE fluid [12,16]. Complement activation in the middle ear mucosa may thus be an indicator of an ongoing persistent inflammation.
The activation of complement was demonstrated with an anti-C3c antibody that recognizes C3 and its breakdown products C3b, iC3b and C3c. Shortly after its formation during complement activation C3b binds to the cell membrane and remains stable maximally for only a few minutes. C3b is inactivated to iC3b by the plasma protease factor I for which the soluble factor H and membrane-associated MCP serve as alternative but essential cofactors. The difference in the distribution iC3b/C3c and C3d apparently depends on the different stages of activation of the C3 molecule. On the mucosal surface C3 deposits are due to recent complement activation and therefore represent mainly iC3b. On the basement membrane iC3b has become further degraded to C3dg and C3d and the C3c epitope has been lost. C3d and C3dg are stable covalently associated C3 activation products that can persist in tissues for prolonged periods. The C3d antibody used (4C2) preferentially recognizes separate C3d and C3dg and only weakly the C3d epitope on C3b/iC3b. Therefore the 4C2 MoAb shows only a relatively weak reactivity with the surface of the epithelium.
Only little deposition of C9 and expression of the terminal complement complex neoepitope was found on the epithelial cell surfaces of the middle ear mucosa. Also, the basement membranes stained only weakly for C9. These results suggest that complement activation does not proceed to the level of MAC on the middle ear epithelial cells. This, in turn, is apparently due to the fact that the complement membrane regulators MCP and CD59 were strongly expressed on the middle ear epithelial cells.
The middle ear mucosa showed strong positive immunostaining for the complement regulators MCP and CD59. DAF has been shown to be strongly expressed on the normal epithelium in the oral cavity, gastrointestinal tract and salivary glands [17,18]. In this study we found only a weak expression of DAF on the surface of the middle ear mucosal epithelium, but the expression got stronger towards the more proximal parts. The vascular endothelium occasionally stained positive for DAF, as has often been observed in other tissues. The inverse relationship between C3 deposition and DAF expression on the epithelium could indicate that the C3 convertase function is more efficiently controlled on the basal than on the surface parts of the epithelium. A reduction in DAF expression could be due to ligand occupancy by C3b or shedding into the fluid phase. Both DAF and MCP protect cells against complement-mediated cytotoxicity [19]. It appears, however, that the turnover rate of C3 is so rapid on the epithelial surface that the C4b/C3b-inactivation promoting function of MCP is not sufficient to prevent formation of active C3 convertases. The evidence thus suggests that the conditions that develop during OME favour amplification of complement activation.
In addition to MCP, CD59 seems to be another main complement regulator in the middle ear mucosa. In normal middle ear mucosa CD59 was found to be expressed by all epithelial cells. In chronic OME patients, the expression was weaker on the outermost surface of the epithelium. This could be due to release of the phospholipid-tailed form of GPI-anchored CD59 to the middle ear effusion or uptake into the MAC complexes. The granular expression of CD59 seen in the middle ear epithelium is similar to that observed in previous studies of adult periodontitis and myocardial infarction [17,20].
In studies on nasal epithelium it has been speculated that an intense complement activation by invading microorganisms may overcome the protection by complement regulators CD59 and DAF and lead to tissue injury and cell death [21]. In the case of middle ear mucosa it appears unlikely that complement activation could cause direct cell death, mostly because of the strong expression of MCP and CD59.
In our material the number of positive bacterial cultures was 22%, which is similar to that reported in other studies [12,14,22]. Even though the majority of effusions in OME seem to be negative in bacterial cultures, the close age relation and increasing evidence of bacteria by polymerase chain reaction (PCR) techniques [22] suggest that OME is usually preceded by a bacterial infection. In an animal model with chinchilla, bacteria persisted in a viable state for weeks after antibiotic treatment in the MEE [23]. The most frequently encountered bacteria in acute OME have been shown to have a high C1q binding capacity [24]. Thus, the bacterial infection could activate the classical complement pathway and the anatomical conditions (tubal insufficiency) could maintain further complement activation.
As shown in this study, an intense complement activation in the middle ear mucosa could contribute to the chronic inflammation in OME. By maintaining the anatomically trapped conditions in the tympanic cavity the tubal dysfunction in chronic OME can further increase complement turnover in the middle ear space. Hence, a vicious circle of self-maintaining or even self-amplifying inflammation in a closed space is created. The cells in the middle ear epithelium, however, seem to be well protected against complement-mediated cell damage.
Acknowledgments
This study was supported by The Academy of Finland, The Sigrid Jusélius Foundation and a state subsidy to The Helsinki University Central Hospital.
REFERENCES
- 1.Klein JO, Tos M, Hussl B, Naunton RF, Ohyama M. Definition and classification, Panel reports 1A. Ann Otol Rhinol Laryngol. 1989;98(Suppl. 139):10. [PubMed] [Google Scholar]
- 2.Gates GA. Adenoidectomy for otitis media with effusion. Ann Otol Rhinol Laryngol. 1994;103:54–57. doi: 10.1177/00034894941030s515. [DOI] [PubMed] [Google Scholar]
- 3.Bluestone CD, Klein JO, Stool SE, Kenna MA. Otitis media in infants and children. 2. Philadelphia: W.B. Saunders Co; 1995. Physiology, pathophysiology and patho-genesis; pp. 17–37. [Google Scholar]
- 4.Bernstein JM, Prellner K, Rynnell-Dagoo B, Sipala P, Palva T. Otitis media: the immunological factor. Arch Otolaryngol Head Neck Surg. 1986;112:203–10. [Google Scholar]
- 5.Ogra PL, Bernstein JM, Yurchak AM, Coppola PR, Tomasi TB. Characteristics of secretory immune system in human middle ear: implication in otitis media. J Immunol. 1974;112:488–95. [PubMed] [Google Scholar]
- 6.Alho OP, Oja H, Koivu M, Sorri M. Chronic otitis media with effusion in infancy. Arch Otolaryngol Head Neck Surg. 1995;121:432–6. doi: 10.1001/archotol.1995.01890040056009. [DOI] [PubMed] [Google Scholar]
- 7.Morgan BP, Meri S. Membrane proteins that protect against complement lysis. Springer Semin Immunopathol. 1994;15:369–96. doi: 10.1007/BF01837366. [DOI] [PubMed] [Google Scholar]
- 8.Meri S, Morgan BP, Davies A, Daniels RH, Olavesen MG, Waldmann H. Human protectin (CD59), an 18,000–20,000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990;71:1–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Rollins SA, Zhao JI, Ninomiya H, Sims PJ. Inhibition of homologous complement by CD59 is mediated by a species-selective recognition conferred through binding to C8 within C5b-8 or C9 within C5b-9. J Immunol. 1991;146:2345–51. [PubMed] [Google Scholar]
- 10.Väkevä A, Laurila P, Meri S. Regulation of complement membrane attack complex formation in myocardial infarction. Am J Pathol. 1993;143:65–75. [PMC free article] [PubMed] [Google Scholar]
- 11.Weisman HF, Bartow T, Leppo MK, et al. Soluble human comple-ment receptor type 1 in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–51. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 12.Meri S, Lehtinen T, Palva T. Complement in chronic secretory otitis media. Arch Otolaryngol. 1984;110:774–8. doi: 10.1001/archotol.1984.00800380004002. [DOI] [PubMed] [Google Scholar]
- 13.Bernstein JM, Byung PH. Chemotactic activity in middle ear effusions. Otolaryngol Head Neck Surg. 1981;89:1007–12. doi: 10.1177/019459988108900625. [DOI] [PubMed] [Google Scholar]
- 14.Yellon RF, Doyle WJ, Whiteside TL, Diven WF, March AR, Fireman P. Cytokines, immunoglobulins, and bacterial pathogens in middle ear effusions. Arch Otolaryngol Head Neck Surg. 1995;121:865–9. doi: 10.1001/archotol.1995.01890080033006. [DOI] [PubMed] [Google Scholar]
- 15.Heimer GV, Taylor CED. Improved mountain for immunofluorescence preparations. J Clin Pathol. 1974;27:254–6. doi: 10.1136/jcp.27.3.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernstein JM, Schenkein HA, Genco RJ, Bartholomew WR. Complement activity in middle ear effusions. Clin Exp Immunol. 1978;33:340–6. [PMC free article] [PubMed] [Google Scholar]
- 17.Rautemaa R, Meri S. Protection of gingival epithelium against complement-mediated damage by strong expression of membrane attack complex inhibitor protectin (CD59) J Dent Res. 1996;75:568–74. doi: 10.1177/00220345960750010901. [DOI] [PubMed] [Google Scholar]
- 18.Medof EM, Walter EI, Rutgers JL, Knowles DM, Nussenzweig V. Identification of the complement decay-accelerating factor (DAF) on epithelium and glandular cells and in body fluids. J Exp Med. 1987;165:848–64. doi: 10.1084/jem.165.3.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lublin DM, Coyne KE. Phospholipid-anchored and transmembrane versions of either decay-accelerating factor or membrane cofactor protein show equal efficiency in protection from complement-mediated cell damage. J Exp Med. 1991;174:35–44. doi: 10.1084/jem.174.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Väkevä A, Laurila P, Meri S. Loss of expression of protectin (CD59) is associated with complement membrane attack complex deposition in myocardial infarction. Lab Invest. 1992;67:608–16. [PubMed] [Google Scholar]
- 21.Varsano S, Frolkis I, Rashkovsky L, Ophir D, Fishelson Z. Protection of human nasal respiratory epithelium from complement-mediated lysis by cell-membrane regulators of complement activation. Am Resp Cell Mol Biol. 1996;15:731–7. doi: 10.1165/ajrcmb.15.6.8969267. [DOI] [PubMed] [Google Scholar]
- 22.Jero J, Virolainen A, Salo P, Leinonen M, Eskola J, Karma P. PCR assay for detecting Streptococcus pneumoniae in the middle ear of children with otitis media with effusion. Acta Otolaryngol (Stockh) 1996;116:288–92. doi: 10.3109/00016489609137843. [DOI] [PubMed] [Google Scholar]
- 23.Post CJ, Aul JJ, White GJ, et al. PCR-based detection of bacterial DNA after antimicrobial treatment is indicative of persistent, viable bacteria in the chinchilla model of otitis media. Am J Otolaryngol. 1996;17:106–11. doi: 10.1016/s0196-0709(96)90005-8. [DOI] [PubMed] [Google Scholar]
- 24.Prellner K. Bacteria associated with acute otitis media have high C1q binding capacity. Acta Path Microbiol Scand. 1980;88:187–90. doi: 10.1111/j.1699-0463.1980.tb00093.x. [DOI] [PubMed] [Google Scholar]