

Abstract
Wegener's granulomatosis (WG) and microscopic polyangiitis are systemic autoimmune diseases characterized by the presence of ANCA in the sera of patients. Little is known about the aetiologic factors and genetic predisposition as well as the pathogenesis of these disease entities. A slightly decreased representation of HLA-DRB1*13 and HLA-DQB1*0603 individuals was observed in our cohort of ANCA-associated systemic vasculitis (AASV) patients compared with controls. In addition, HLA-DRB1*04 individuals were over-represented in a subgroup of patients with WG in end-stage renal disease as a result of renal vasculitis. In order to identify other genes relevant for these diseases, we investigated highly polymorphic markers in the vicinity of several immunorelevant genes, i.e. tumour necrosis factor (TNF)α, IL-2, IL-5 receptor α (IL-5RA), in a group of 102 patients with AASV and compared the representation with controls. Furthermore, functional polymorphisms were directly analysed in the promotor region of TNFα as well as in the coding region of the FcγIIRA genes. Polymorphisms of the TNFα promotor (TNF-308) as well as in the FcγIIRA gene were excluded as risk factors for the disease in our cohort. No major phenotype distribution differences were observed between patients and controls for the IL-2 and IL-5RA microsatellites. Most importantly, several haplotypes on chromosome 6p appeared strongly associated with proteinase 3 (PR3)-ANCA+ AASV. Thus, as in other autoimmune diseases, different predisposing factors play differential aetiopathogenic roles in various groups of AASV patients.
Keywords: genetic predisposition, anti-neutrophil cytoplasmic antibody, Wegener's granulomatosis, HLA, tumour necrosis factor-alpha
INTRODUCTION
Major features of Wegener's granulomatosis (WG) and microscopic polyangiitis are inflammatory lesions of the upper and lower airways and pauciimmune glomerulonephritis with and without granuloma formation. The disease manifestations result from small vessel vasculitis. A characteristic feature for this group of diseases is the occurrence of ANCA, i.e. IgG autoantibodies against proteinase 3 (PR3), myeloperoxidase (MPO) and rarely azurozidin (AZU) as well as bacterial permeability-increasing protein (BPI). All autoantigens are located in the primary azurophilic granules of monocytes, polymorphonuclear granulocytes (PMN) [1] and on the surface of tumour necrosis factor-alpha (TNF-α)-primed PMN [2]. Several properties of ANCA have been described. PR3-ANCA promote the adhesion of PMN on human blood vessel walls via induction of E-selectin and vascular cell adhesion molecule-1 (VCAM-1) on endothelial cells [3,4] and of Mac-1 (CD11b/CD18; granulocyte adhesion molecule) on PMN [5]. Secondly, the binding of PR3-ANCA as well as MPO-ANCA to PMN activated via CD18, leads to release of free oxygen radicals and proteases via concurrent binding of PR-3 and FcγIIRA, both expressed on the PMN cell surface [6].
The search for associations of HLA class II alleles with WG has produced conflicting results. Either the associated HLA antigens varied in different studies [7–11] or disease associations were not demonstrated [12–15]. Due to the very low prevalence, the inheritance pattern of WG is also not clear. Furthermore, the association of WG with the defective (Z) allele of the proteinase inhibitor (Pi) of the α1-antitrypsin (α1-AT) locus, a potent PR3 inhibitor, evidences relevant genetic factors other than the HLA region [16–18]. Allelic variants of additional immunorelevant proteins may alter the function or the ligand–receptor interaction and may contribute in combination with other exogenous or endogenous factors to the manifestation of autoimmune disease.
Compared with Mendelian diseases, multifactorial traits are far more complex to investigate using molecular genetic techniques. Different strategies are used in the search for susceptibility regions and candidate genes for complex traits [19–21]. The aim of this study was to investigate polymorphisms of several immunorelevant candidate genes in a German cohort of patients with ANCA-associated systemic vasculitis (AASV). Inflammation processes are driven by a multitude of cytokines, some of which are known to be proinflammatory. TNF-α is a cytokine responsible for PMN priming, sensitizing them for further activation, e.g. by PR3-ANCA. IL-2 also exhibits strong proinflammatory activity. Expressed on B cells, the IL-5 receptor α (IL-5RA) is required for development of antibody-secreting cells. Therefore these genes were among the prime candidates for genetic factors contributing to the risk for developing AASV. Known polymorphisms in the TNFα promotor and an exonic polymorphism in the FcγIIRA gene were typed directly. The investigation of putative polymorphisms in the IL-2, IL-5RA and TNFα genes was performed indirectly, using either intragenic or neighbouring microsatellite markers, because one or several alleles of the respective microsatellite are supposed to be in linkage disequilibrium with the putative coding polymorphism. Potential genetic predisposition factors were evaluated also for the different subgroups of AASV patients.
PATIENTS AND METHODS
Patients and controls
A total of 102 patients with AASV were included in this study. PR3-ANCA were detectable in 76 patients and 26 patients had MPO-ANCA. All patients gave informed consent, originated from Bavaria and were diagnosed and treated in the Departments of Medicine (Medizinische Klinik and Poliklinik, Klinikum Innenstadt, Medizininische Klinik I, Klinikum Großhadern, Ludwig-Maximilians-University, München; Department of Medicine IV, Städtisches Krankenhaus Harlaching, München; Department of Medicine, Klinikum Hohe Warte, Bayreuth; Department of Nephrology, St Joseph Hospital, Regensburg, Germany).
Serum PR3- and MPO-ANCA were demonstrated by indirect immunofluorescence (IIF) using ethanol-fixed neutrophils and their reactivity with PR3 or MPO was confirmed in ELISAs specific for PR3 and MPO (Elias, Freiburg, Germany), respectively. The patients were classified as having small vessel vasculitis. Crescent and necrotizing glomerulonephritis was considered to be part of the vasculitic spectrum. The diagnosis of ANCA vasculitis was determined in accordance with the definitions for systemic vasculitides by the Chapel Hill consensus conference (CHC) [22] and the classification criteria defined by the American College of Rheumatology (ACR) [23]. As controls, genotype data from a ‘southern German’ group of healthy donors were provided by E.A.
Clinical data
In order to compare groups of patients with regard to systemic dissemination of the disease, the involvement of different organ systems was recorded. The extent of disease manifestation was determined in accordance with the definitions of the disease extension index (DEI [24]) representing an extended ear/nose/throat/lung/kidney (ELK) classification [25].
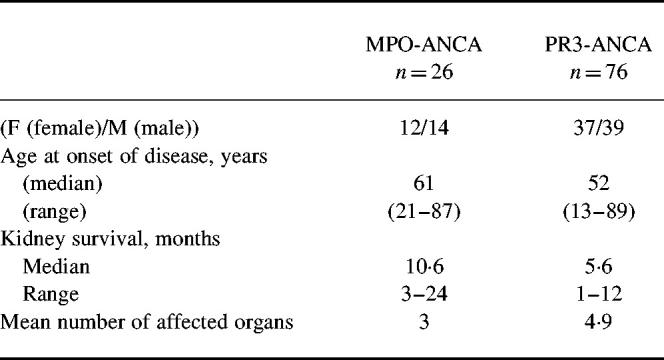
Symptoms and signs compatible with vasculitic involvement in the following organ systems were recorded: ear, nose and throat (including larynx and paranasal sinuses); lung; kidney; eyes; skin; joints and muscles; heart, gastrointestinal tract; liver; peripheral/central nervous system; others (e.g. fever, weight loss). Upper respiratory tract involvement was mainly based on symptoms (e.g. epistaxis), sinus x-rays and nasal biopsies. Lung involvement was judged from x-ray reports, fibroscopic bronchial biopsy samples or open lung biopsy. Kidney involvement was documented by biopsy, the follow up was based on changes in serum creatinine concentration and/or urinary findings (Table 1). According to the extent of the disease the PR3-ANCA+ subgroup of patients with end stage renal disease (ESRD PR3-ANCA) or patients who died from the disease was analysed separately (n = 32).
Table 1.
Baseline characteristics of myeloperoxidase (MPO)-ANCA+ and proteinase 3 (PR3)-ANCA+ vasculitis patients
DNA preparation, HLA typing, microsatellite analyses and allele specific (ASO) hybridization
DNA was extracted from peripheral blood according Miller et al. [26]. Typing of HLA-DR and HLA-DQ has been detailed previously [27]. Microsatellite analyses of IL-2, IL-5RA and TNFa were performed using polymerase chain reaction (PCR) amplification with previously described primers [28,29]. TNFα promotor region polymorphism (G → A transition at position 308) was analysed according to McGuire et al. [30]. The FcγIIRA polymorphism was analysed using PCR primers and conditions described by Osborne et al. [31].
Statistical analysis
Statistical analyses were performed for both groups of AASV patients as well as for the ESRD subgroup of patients with PR3-ANCA. Statistical significance was evaluated by the χ2 test or Fisher's exact test. The P values have been corrected for the number of comparisons made according to Svejgaard & Ryder (P = uncorrected, Pc = corrected [32]). Relative risk (RR) was calculated as described [33]. Linkage disequilibria (LD) and probability values were calculated with the on-line version of LINKDOS [34]. Haplotype frequencies were estimated with the software Arlequin [35].
RESULTS
HLA-DRB1 and DQB1 typing
Patients with AASV and the healthy controls were typed for HLA-DRB1 alleles. The DRB1 phenotype frequencies for the PR3- and MPO-ANCA+ patients and controls are given in Fig. 1. Decreased occurrence of DRB1*13 antigens was observed among PR3-ANCA+ patients compared with controls (12.5% versus 25.5%, RR = 0.4, P < 0.05). The comparison of the overall phenotype frequencies of the DRB1*13/14 antigens in PR3-ANCA+ patients and controls reached statistical significance after correction for multiple comparisons (RR = 0.3, Pc < 0.05). The frequency of the DRB1*04 type was elevated in the ESRD subgroup of PR3-ANCA+ compared with the control group (53.5% and 26.1%, RR = 3.24, P < 0.005).
Fig. 1.
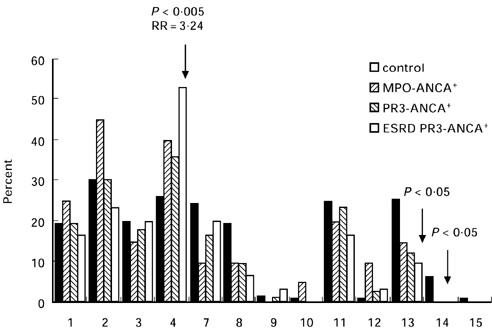
Phenotype frequencies of HLA-DRB1 in controls, myeloperoxidase (MPO)-ANCA+, proteinase 3 (PR3)-ANCA+ and the end stage renal disease (ESRD) subgroup of ANCA-associated systemic vasculitis (AASV) patients. Indicated P values represent significant differences of patient groups versus controls.
The phenotype frequencies of the HLA-DQB1 alleles are shown in Fig. 2. The only statistically significant difference between both AASV groups and the control group was a significant decrease of the DQB1*0603 phenotype within the PR3-ANCA+ patient group (3% and 16.8%, RR = 0.2, P < 0.005).
Fig. 2.
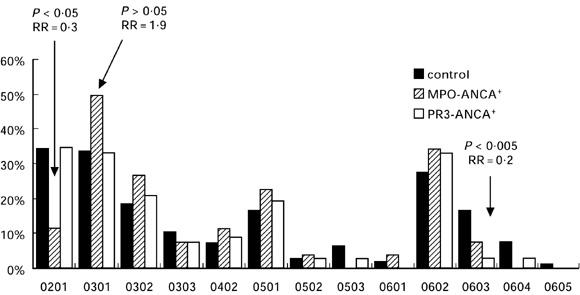
Phenotype frequencies of HLA-DQ in controls, myeloperoxidase (MPO)-ANCA+ and proteinase 3 (PR3)-ANCA+ ANCA-associated systemic vasculitis (AASV) patients.
Typing of the IL-2 and IL-5RA microsatellites and the FcγIIRA polymorphism
Phenotype frequencies were compared between healthy controls and patients for the IL-2 and IL-5RA microsatellites, respectively. No significant differences were found between the controls and the PR3- as well as the MPO-ANCA+ groups (data not shown). Genotyping of the exonic polymorphism in the FcγIIRA gene in the AASV and control groups revealed similar allele and genotype distributions. No significant differences were observed between the controls and both AASV groups (data not shown).
Typing of TNFa microsatellite and TNF-308 promotor polymorphisms
The phenotype frequencies of the TNFa microsatellite are shown in Fig. 3. The phenotype frequency of allele 6a and 11a is elevated in patients with PR3-ANCA+ compared with the controls (6a: 32% and 18.2%, RR = 2.1, P < 0.05; 11a: 45.3% and 29.87%, RR = 1.9, P < 0.05). For the same locus the TNFα promotor polymorphism was investigated in our AASV patients and in the healthy control group. No difference was detected between the allele and genotype frequencies in the promotor region of the TNFα gene between patients and controls (data not shown).
Fig. 3.
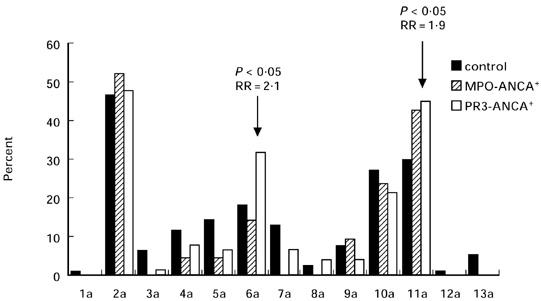
Phenotype frequencies of the TNFa microsatellite alleles in controls, myeloperoxidase (MPO)-ANCA+ and proteinase 3 (PR3)-ANCA+ ANCA-associated systemic vasculitis (AASV) patients.
Linkage disequilibria analysis between TNFa microsatellite, HLA-DRB1 and HLA-DQB1 loci
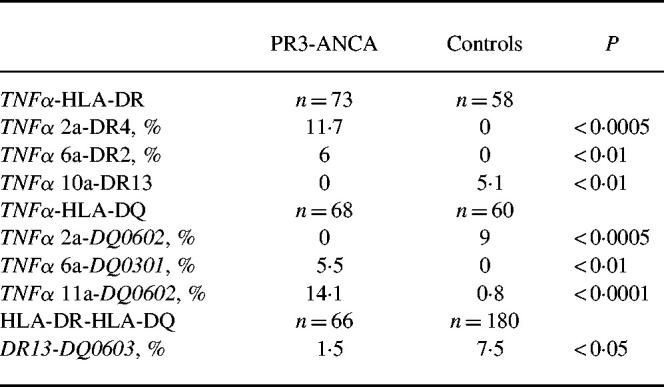
LD analysis was performed between the TNFa microsatellite, the HLA-DRB1 and the DQB1 loci. TNFa and DRB1 as well as TNFa and DQB1 loci appeared in stronger LD in AASV patients compared with controls (P < 0.0005 versus P > 0.05). This result stands in contrast to the situation of the DRB1 and DQB1 loci, which were in LD in both cohorts. Several significant haplotype frequency differences were evident between the TNFa microsatellite and HLA-DRB1/DQB1 in the PR3-ANCA+ patient versus the control group. Haplotype analyses was not performed for the MPO-ANCA+ group due to insufficient numbers of patients. The main differences in presumable haplotype frequencies between the PR3-ANCA+ patients and the control groups are shown in Table 2. Although there were several significant haplotype frequency differences between the TNFa microsatellite and the DRB1 locus as well as the TNFa microsatellite and the DQB1 locus, these haplotypes apparently did not encompass this entire region.
Table 2.
Haplotype frequencies of the TNFa microsatellite, the HLA-DR and the HLA-DQ types in proteinase 3 (PR3)-ANCA+ patients and controls
DISCUSSION
Disease associations of WG with HLA antigens were observed several decades ago, albeit with contradictory results. Our results support the findings of Hagen et al. [11], i.e. DRw6 (subtype DRB1*13) tends to occur with reduced frequencies in patients with WG. These authors presented a negative ‘HLA–DR13DR6’ association with AASV in a group of 224 patients compared with a control cohort comprising more than the 10-fold number of individuals. Our data support an association of the HLA class II locus and a possible ‘protective’ effect of DRB1*13/14 antigens. The interpretation of this ‘protective’ effect of the DRB1*13/14 and DQB1*0603 types in the extended haplotype DRB1*13-DQB1*0603 and the functional relevance for the induction of autoreactive T cells is not straightforward. DRB1*13/14 and/or DQB1*0603 antigens, when presenting antigens from azurophilic granules, may modulate the response of autoreactive T cells. Carefully designed studies with further differentiation of the DRB1*13 antigens comprising larger patient cohorts and matched controls must be performed to confirm a negative association of AASV with the DRB1*13/14 antigen in our population. To identify predisposing genetic factors which increase the risk of disease development we investigated additional relevant genes in the (auto)immune response of AASV. For loci encoding the proinflammatory molecules IL-2 as well as IL-5RA, no association between PR3-ANCA manifestation and these genes was evident.
PR3- and MPO-ANCA have been shown to induce the respiratory burst in PMN and monocytes. A mechanism by which ANCA activate the signal transduction system from the cell surface leading to release of free oxygen radicals is supposed to be mediated through FcγIIRA [6]. FcγIIRA genes express two alleles codominantly, R131 and H131. Both alleles differ in the ability to bind IgG2 and IgG3. The H131 allele is the only Fc receptor recognizing human IgG2 efficiently [36]. Heterozygous and homozygous carriers of the R131 allele were shown to be at higher risk of bacterial infections due to disturbed phagocytic functions of PMN and monocytes [37–39]. Additionally, the R131 allele is associated with nephritis in systemic lupus erythematosus (SLE) [40]. PMN H131/H131 and R131/R131 homozygous individuals differ by more than three-fold in the production of free oxygen radicals after binding ANCA to their ligand [41]. The authors concluded that FcγIIRA alleles may represent risk factors influencing the release of free oxygen radicals in inflammatory processes and tissue injury. The data in our cohorts as well as another study on 147 WG patients published recently [42] exclude an association of the FcγRIIA polymorphism and the manifestation of AASV. Local TNF-α production was found to be increased in renal biopsies of ANCA+ glomerulonephritis and the TNF-α concentration correlated with the activity of the disease [43]. The TNFα gene is situated in the class III region of the HLA complex on chromosome 6. The TNF-308 ( A) allele of the promotor is part of an extended haplotype in the HLA region (including the DRB1*03 antigen) and was shown to be associated with increased TNF-α production and with other inflammatory diseases such as rheumatoid arthritis, asthma and malaria [44–46]. To evidence a possible association of the genetically determined over-production of TNF-α and its subsequent over-stimulation of PMN with clinical manifestations of AASV, this polymorphism was typed in our cohorts. Our data support a recently published study excluding disease association with the promotor polymorphism TNF-308 in the patients [47]. Furthermore, we genotyped a polymorphic microsatellite in the vicinity of the TNFα gene. In contrast to the TNFα promotor polymorphism, in this case the frequencies differed between the patient and the control groups. Interestingly, we found an increased representation of allele 11a. The high phenotype frequencies of the allele 11a as well as the over-representation especially of the TNFa 2a/DRB1*04 and TNFa 11a/DQB1*0602 extended haplotypes in vasculitis patients with ESRD, together with a lack of a common haplotype between the TNFα, HLA-DR and DQ loci, indicate however that another functional polymorphism might be located in the vicinity, contributing to the pathogenesis and especially to severe courses of WG. HLA typing as well as other markers in the MPO-ANCA+ patient group show similarities to as well as a few differences from the PR3-ANCA+ group. However, no statistical analysis could be performed because of the small number of individuals in this group.
In addition to the confirmed association of WG with the PiZ allele of the α1-AT gene and the protective effect of the DRB1*13/14 allele, no major genetic predisposing factors are known to play a role in WG and in AASV. Our data suggest that there are other polymorphisms located in the HLA region as well as in other genomic regions conferring susceptibility to AASV and relevant to the clinical course of the disease.
Acknowledgments
The authors thank Professors D. Schlöndorff, D. Seybold, F. E. Scherberich and W. Samtleben for enabling us to study patients treated in their Departments of Medicine.
REFERENCES
- 1.Raltson DR, Marsh CB, Lowe MP, Wewers MD. Antineutrophil cytoplasmic antibodies induce monocyte IL-8 release. J Clin Invest. 1997;100:1416–24. doi: 10.1172/JCI119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falk RJ, Terell RS, Charles LA, Jenette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayet W-J, Meyer zumBüschenfelde K-H. Antibodies to proteinase 3 increase adhesion of neutrophils to human endothelial cells. Clin Exp Immunol. 1993;94:440–6. doi: 10.1111/j.1365-2249.1993.tb08215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayet W-J, Scharting A, Orth T, Duchmann R, MeyerzumBüschenfelde K-H. Antibodies to proteinase 3 mediate expression of vascular cell adhesion molecule-1 (VCAM-1) Clin Exp Immunol. 1996;103:259–67. doi: 10.1046/j.1365-2249.1996.d01-626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson PA, Alexander HD, McMillan SA, Maxwell AP. Up-regulation of the granulocyte adhesion molecule Mac-1 by autoantibodies in autoimmune vasculitis. Clin Exp Immunol. 1997;107:513–9. doi: 10.1046/j.1365-2249.1997.d01-956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mulder AHL, Heeringa P, Brouwer E, Limburg PC, Kallenberg CGM. Activation of granulocytes by anti-neutrophil antibodies (ANCA): a FcγRII-dependent process. Clin Exp Immunol. 1994;98:270–8. doi: 10.1111/j.1365-2249.1994.tb06137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elkon KB, Sutherland DC, Rees AJ, Hughes GR, Batchelor JR. HLA antigen frequencies in systemic vasculitis: increase in HLA-DR2 in Wegener's granulomatosis. Arthritis Rheum. 1983;26:102–5. doi: 10.1002/art.1780260118. [DOI] [PubMed] [Google Scholar]
- 8.Katz P, Alling DW, Haynes BF, Fauci AS. Association of Wegener's granulomatosis with HLA-B8. Clin Immunol Immunopathol. 1979;14:268–70. doi: 10.1016/0090-1229(79)90150-8. [DOI] [PubMed] [Google Scholar]
- 9.Spencer SJW, Burns A, Gaskin G, Pusey CD, Rees AJ. HLA class II specificities in vasculitis with antibodies to neutrophil cytoplasmic antigens. Kidney Int. 1992;41:1059–63. doi: 10.1038/ki.1992.161. [DOI] [PubMed] [Google Scholar]
- 10.Papiha SS, Murty GE, Ad Hia A, Mains BT, Venning M. Association of Wegener's granulomatosis with HLA antigens and other genetic markers. Ann Rheum Dis. 1992;51:246–8. doi: 10.1136/ard.51.2.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagen EC, Stegmann CA, D'amaro J, et al. Decreased frequency of HLA-DR13DR6 in Wegener's granulomatosis. Kidney Int. 1995;48:801–5. doi: 10.1038/ki.1995.353. [DOI] [PubMed] [Google Scholar]
- 12.Strimlan CV, Taswell HF, Kueppers F, Deremee RA, McDonald TJ. HLA-A antigens of patients with Wegener's granulomatosis. Tissue Antigens. 1978;11:129–31. doi: 10.1111/j.1399-0039.1978.tb01236.x. [DOI] [PubMed] [Google Scholar]
- 13.Beigel A, Lehmann H, Westphal E. The spectrum of histocompatibility antigens (HLA) in Wegener's granulomatosis. Arch Otorhinolaryngol. 1981;233:157–60. doi: 10.1007/BF00453639. [DOI] [PubMed] [Google Scholar]
- 14.Murty GE, Mains BT, Middleton D, Maxwell AP, Savage DA. HLA antigen frequencies and Wegener's granulomatosis. Clin Otolaryngol. 1991;16:448–51. doi: 10.1111/j.1365-2273.1991.tb01037.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Jayne DRW, Zhao MH, Lockwood CM, Oliveira DBG. Distribution of MHC class II alleles in primary systemic vasculitis. Kidney Int. 1995;47:294–8. doi: 10.1038/ki.1995.37. [DOI] [PubMed] [Google Scholar]
- 16.Elzouki ANY, Segelmark M, Wieslander J, Eriksson S. Strong link between the alpha1-antitrypsin PiZ allele and Wegener's granulomatosis. J Intern Med. 1994;236:543–8. doi: 10.1111/j.1365-2796.1994.tb00842.x. [DOI] [PubMed] [Google Scholar]
- 17.Esnault VLM, Testa A, Audrain M, et al. Alpha1-antitrypsin genetic polymorphism in ANCA-positive systemic vasculitis. Kidney Int. 1993;43:1329–32. doi: 10.1038/ki.1993.186. [DOI] [PubMed] [Google Scholar]
- 18.Segelmark M, Elzouki AN, Wieslander J, Eriksson S. The PiZ gene of α1-antitrypsin as a determinant of outcome in PR3-ANCA-positive vasculitis. Kidney Int. 1995;18:844–50. doi: 10.1038/ki.1995.360. [DOI] [PubMed] [Google Scholar]
- 19.Sobell JL, Heston LL, Sommer SS. Delineation of genetic predisposition to multifactorial disease: a general approach on the treshold of feasibility. Genomics. 1992;12:1–6. doi: 10.1016/0888-7543(92)90398-c. [DOI] [PubMed] [Google Scholar]
- 20.Vyse TJ, Todd JA. Genetic analysis of autoimmune diseases. Cell. 1996;85:311–8. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 21.Goldgar DE, Easton DF. Optimal strategies for mapping complex diseases in the presence of multiple loci. Am J Hum Genet. 1997;60:1222–32. [PMC free article] [PubMed] [Google Scholar]
- 22.Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides; Proposal of an international consensus conference. Arthritis Rheum; 1994. pp. 187–92. [DOI] [PubMed] [Google Scholar]
- 23.Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum. 1990;33:1101–7. doi: 10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 24.DeRemee RA, McDonald TJ, Harrison EG, Coles DT. Wegener's granulomatosis. Anatomic correlates, a proposed classification. Mayo Clin Proc. 1976;51:777–81. [PubMed] [Google Scholar]
- 25.Reinhold-Keller E, Kekow J, Schnabel A, et al. Influence of disease manifestation and antineutrophil cytoplasmic antibody titer on the response to pulse cyclophosphamide therapy in patients with Wegener's granulomatosis. Arthritis Rheum. 1994;37:919–24. doi: 10.1002/art.1780370622. [DOI] [PubMed] [Google Scholar]
- 26.Miller SA, Dykes DD, Poleski HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haas JP, Kimura A, Andreas A, et al. Polymorphism in the upstream regulatory region DQA1 genes and DRB1, QAP, DQA1, and DQB1 haplotypes in the German population. Hum Immunol. 1994;39:31–40. doi: 10.1016/0198-8859(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 28.Epplen C, Frank G, Gomolka M, Nagy M, Albert E, Nürnberg P, Epplen JT. Dinucleotide repeat polymorphism in the IL2 and ILR5RA genes. Hum Mol Genet. 1994;3:679. doi: 10.1093/hmg/3.4.679-a. [DOI] [PubMed] [Google Scholar]
- 29.Gomolka M, Menninger H, Saal JE, et al. Immunoprinting: various genes are associated with increased risk to develop rheumatoid arthritis in different groups of adult patients. J Mol Med. 1995;73:19–29. doi: 10.1007/BF00203615. [DOI] [PubMed] [Google Scholar]
- 30.McGuire W, Hill AVS, Allsopp CEM, Greenwood BM, Kwiatkowski D. Variation in the TNFα promotor region associated with susceptibility to cerebral malaria. Nature. 1994;371:508–10. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 31.Osborne JM, Chacko GW, Brandt JT, Anderson CL. Ethnic variation in frequency of an allelic polymorphism of human FcγIIRA determined with allele specific oligonucleotide probes. J Immunol Methods. 1994;173:207–17. doi: 10.1016/0022-1759(94)90299-2. [DOI] [PubMed] [Google Scholar]
- 32.Svejgaard A, Ryder LP. HLA and disease associations: detecting the strongest association. Tissue Antigens. 1994;43:18–27. doi: 10.1111/j.1399-0039.1994.tb02291.x. [DOI] [PubMed] [Google Scholar]
- 33.Svejgaard A, Platz P, Ryder LP. HLA and disease 1982—a survey. Immunol Rev. 1982;70:193–18. doi: 10.1111/j.1600-065x.1983.tb00715.x. [DOI] [PubMed] [Google Scholar]
- 34.Garnier-Gere P, Dillmann C. A computer program for testing pairwise linkage disequilibria in subdivided populations. J Heredity. 1992;83:239. doi: 10.1093/oxfordjournals.jhered.a111204. [DOI] [PubMed] [Google Scholar]
- 35.Schneider S, Kueffer J-M, Roessli D, Excofier L. Geneva: Genetics and Biometry Lab, Dept.of Anthropology, University of Geneva; 1997. Arlequin: a software for population genetic data analysis, Version 1.1. [Google Scholar]
- 36.Salmon JE, Edberg JC, Kimberly RP. Allelic polymorphism of human Fc receptor IIA and Fc receptor IIIB. Independent mechanisms for differences in human phagocyte function. J Clin Invest. 1992;89:1274–81. doi: 10.1172/JCI115712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stuart SG, Trounstine M, Vaux D, Koch T, Martens C, Mellmann I, Moore KM. Isolation and expression of cDNA clones encoding a human receptor for IgG (Fc-RII) J Exp Med. 1987;166:1668–84. doi: 10.1084/jem.166.6.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bredius RGM, Derkx BHF, Fijen CAP, et al. Fcγ receptor IIa (CD32) polymorphism in fulminant meningococcal septic shock in children. J Infect Dis. 1994;170:848–53. doi: 10.1093/infdis/170.4.848. [DOI] [PubMed] [Google Scholar]
- 39.Sanders LAM, Feldman RG, Voorhorst-Ogink MM, DeHaas M, Rijkers GT, Capel PJA, Van de Winkel JGJ. Human immunoglobulin G (IgG) Fc receptor IIA (CD32) polymorphism and IgG2-mediated bacterial phagocytosis by neutrophils. Infect Immunol. 1995;63:73–81. doi: 10.1128/iai.63.1.73-81.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salmon JE, Millard S, Schachter LA, et al. Fcγ-RIIA alleles are heritable risk factors for lupus nephritis in African Americans. J Clin Invest. 1996;97:1348–54. doi: 10.1172/JCI118552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porges AJ, Redecha PB, Taylor Kimberly W, Csernok E, Gross WL, Kimberly RP. Anti-neutrophil cytoplasmic antibodies engage and acti-vate human neutrophils via FcγRIIa. J Immunol. 1994;153:1271–80. [PubMed] [Google Scholar]
- 42.Edberg JC, Wainstein E, Wu J, et al. Analysis of FcγRII gene polymorphisms in Wegener's granulomatosis. Exp Clin Immunogenet. 1997;14:183–95. [PubMed] [Google Scholar]
- 43.Noronha IL, Kruger C, Andrassy K, Ritz E, Waldherr R. In situ production of TNF-alpha, IL-1 beta and IL-2R in ANCA-positive glomerulonephritis. Kidney Int. 1993;43:682–92. doi: 10.1038/ki.1993.98. [DOI] [PubMed] [Google Scholar]
- 44.di Giovine FS, Nuki G, Duff GW. Tumour necrosis factor in synovial exudates. Ann Rheum Dis. 1988;47:768–72. doi: 10.1136/ard.47.9.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moffat MF, Cookson WOCM. Tumor necrosis factor haplotypes and asthma. Hum Mol Genet. 1997;6:551–4. doi: 10.1093/hmg/6.4.551. [DOI] [PubMed] [Google Scholar]
- 46.Kwiakowski D, Hill AV, Sambou I, et al. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 1990;336:1201–4. doi: 10.1016/0140-6736(90)92827-5. [DOI] [PubMed] [Google Scholar]
- 47.Mascher B, Schmitt W, Csernok E, Tatsis E, Reil A, Gross WL, Seyfarth M. Polymorphisms in the tumor necrosis factor genes in Wegener's granulomatosis. Exp Clin Immunogenet. 1997;14:226–33. [PubMed] [Google Scholar]