

Abstract
The costimulatory molecule CD28 is expressed on almost all CD4+ T cells, but on only a portion of CD8+ T cells in healthy human adults. αβ T cells may thus be divided into three phenotypically and functionally different subsets: CD4+, CD8+CD28+ and CD8+CD28−. Using peripheral blood lymphocytes from six healthy adults, we have studied the T cell receptor (TCR) repertoire within these subsets by analysis of the distribution of lengths of the complementarity determining region 3 (CDR3) of the beta variable (BV) transcripts and flow cytometric analysis of TCR Vβ usage. Expanded CDR3 lengths were identified in 86% of BV families within CD8+CD28− T cells, but in only 4% within CD4+ T cells, and 35% within CD8+CD28+ T cells (P < 0.01). When sequenced, the majority of expanded peaks were found to be dominated by single clones. Identical expanded clones were found within both CD8+CD28+ and CD8+CD28− subsets, consistent with the belief that CD8+CD28− T cells descend directly from CD8+CD28+ T cells. Greatly expanded CD28− clones were found within both CD8+ and CD4+ subsets and persisted at the same magnitude for up to 4.5 years of observation. The finding of a small proportion of cells expressing Ki-67 showed that some of these clonally expanded cells were in the active stages of the cell cycle, but few of the cells expressed activation markers CD69, CD25, CD71 or CD122. One likely explanation for the persistence of expanded peripheral lymphocyte populations in healthy individuals is the presence of persistent antigen.
Keywords: T cell, CD8, CD28, CDR3 length analysis, Vβ family
INTRODUCTION
T cell response is determined by the T cell receptor (TCR). The most common type of TCR is the αβ TCR, which recognizes antigenic peptide presented by the MHC. Each T cell or clone of cells possesses a unique TCR. Specificity is determined by the sequence within the hypervariable regions, in particular the complementarity determining region 3 (CDR3) [1,2]. The activation of naive T cells is believed to require two independent signals, recognition of antigen by the TCR, and costimulation. The most important costimulatory receptor on the T cell surface is the CD28 molecule [3]. Absence of proper costimulation may result in T cell anergy [4,5]. Nonetheless, in a healthy adult up to 30% or more of circulating CD8+ T cells (but very few CD4+ T cells) lack CD28 [6].
Clonally expanded populations within the CD8+ subset observed in elderly individuals were believed to be the cellular counterpart to benign monoclonal gammapathy [7]. Similar expansions have also been described in healthy adults [8–14]. CD8+CD28− T cells were found to be unresponsive to TCR or CD3 stimulation in vitro [6], and their functional role in vivo remains largely unknown. In HIV infection, the number of CD8+CD28− cells in blood rises dramatically. During the asymptomatic phase of infection, when the CD8+ T cell subset is greatly expanded, CD8+CD28− T cells may account for the entire rise in CD8+ cells [6]. In vitro, CD8+CD28− cells show a cytotoxic capacity for HIV-1-infected cells [15].
To expand our knowledge of the ontogeny and functional role of CD8+CD28− T cells, we have compared the TCR repertoire of this subset with that observed within CD8+CD28+ and CD4+ T cells in six healthy adults. The results are consistent with the notion that CD8+CD28− T cells are end-stage cells derived from CD8+CD28+ precursors.
SUBJECTS AND METHODS
Study participants
Blood samples were obtained from laboratory workers and healthy blood donors to the Rikshospitalet and Red Cross Blood Bank in Oslo. T cell subsets from six persons (mean age 43.9 years, range 19–61 years) were characterized.
Monoclonal antibodies
Anti-CD8 MoAb ITI-5C2 was produced in our laboratory by G. Gaudernack. Anti-CD4 MoAb 66.1 and anti-CD28 MoAb 9.3 were gifts from J. A. Hansen (Fred Hutchinson Cancer Center, Seattle, WA), and anti-CD28 MoAb 15E8 from R. A. W. van Lier (Central Laboratory of the Netherlands Red Cross Blood Transfusion Service, Amsterdam, The Netherlands). These MoAbs were coated onto M-450 Dynabeads (Dynal, Oslo, Norway) as described previously [16] and used for positive and negative selection of lymphocytes. The MoAbs for the variable β-chain (Vβ) phenotype were unconjugated or FITC-conjugated 1C1 (anti-Vβ5.2 + 3), 16G8 (anti-Vβ8), JU74 (anti-Vβ13.3) (Endogen, Woburn, MA); IMMU546 (anti-Vβ22) (Immunotech, Marseille, France); anti-TCRαβ MoAb BMA031 (Behringwerke Diagnostica, Marburg, Germany); anti-αβ TCR (Pharmingen, San Diego, CA). The MoAbs for other cell surface markers were FITC-conjugated Annexin-V, anti-HLA-DR, anti-CD25 (anti-IL-2R), Leu-18 (anti-CD45RA), Leu-7 (anti-CD57), anti-CD71 (anti-transferrin receptor), and PE-conjugated Leu-28 (anti-CD28), Leu-45RO (anti-CD45RO), Leu-23 (anti-CD69), anti-CD122 (anti-IL-2Rβ) (Becton Dickinson Immunocytometry Systems, San Jose, CA); PE-conjugated anti-LFA-1 (anti-CD11A), anti-Fas (anti-CD95 clone DX2), and isotypic control MoAb (PharMingen); MoAb 60.1 (anti-CD11B; a gift from P. G. Beatty (Fred Hutchinson Cancer Center)); FITC-conjugated Ki-67 (Immunotech). Second layer reagent for unconjugated MoAb was FITC- or PE-conjugated polyclonal goat anti-mouse (Pharmingen or Tago Inc. (Burlingame, CA)).
Preparation of cells and flow cytometry
Peripheral blood mononuclear cells (PBMC) were obtained by gradient centrifugation (Lymphoprep; Nycomed, Oslo, Norway). CD4+ and CD8+ cells were positively selected using anti-CD4 and anti-CD8 coated Dynabeads, four beads per target cell as described previously [16]. Cells were detached from beads by incubation for 1 h at room temperature with goat anti-mouse Fab antiserum (DetachAbead; Dynal), washed once and re-suspended in RPMI 1640 (Gibco LT, Gaithersburg, MD) supplemented with 10% human pool serum (HPS) (medium). CD8+ cells were incubated for 45 min at 4°C with Dynabeads coated with anti-CD28 MoAb 9.3 (10/cell) and anti-CD28 MoAb 15E8 (4/cell). Beads and rosetted cells were isolated on a magnet and washed three times; remaining cells were always > 94% CD8+CD28−. CD4+ T cells were not further subdivided due to the small number of CD4+CD28− T cells in the study subjects and in most healthy individuals [6]. For CDR3 length analysis, CD8+CD28− cells were pelleted from single-cell suspension and CD8+CD28+ cells as rosettes. CD4+ T cells were pelleted as single-cell suspensions or as rosettes. Fractions of 1.0 × 106 cells were stored as dry pellets at −70°C in siliconized, autoclaved microcentrifuge tubes. For use as positive controls, PBMC were activated with phytohaemagglutinin (PHA) at 2 μg/ml (Murex Diagnostics Ltd, Dartford, UK) and IL-2 (Amersham, Cleveland, OH) at 5 U/ml for 3 or 6 days.
Staining with MoAb was always performed on 2–3 × 105 fresh cells. Cells were incubated with directly conjugated MoAb at the appropriate concentration in 50 μl medium for 15 min at 4°C, washed twice, and fixed in 1% paraformaldehyde. For Annexin V, cells were washed once in PBS without HPS before staining and twice in Binding Buffer (Pharmingen) after staining. For staining with unconjugated primary MoAb, cells were incubated with MoAb at the appropriate concentration for 30 min at 4°C, washed twice, incubated with FITC- or PE-conjugated goat anti-mouse antibodies for 30 min at 4°C, and washed again. Cells were then incubated with freshly thawed mouse serum at 1:10 for 10 min at 4°C to block all free binding sites on the goat anti-mouse antibodies and then incubated with directly conjugated MoAb as above.
After staining with cell surface MoAb, cells for staining with Ki-67 or control MoAb were washed once and re-suspended in 100 μl PBS without HPS. They were then held in 1 ml Ortho Permeafix solution (Ortho Diagnostic Systems Inc., Raritan, NJ) for 30 min at room temperature, washed once with PBS–2% HPS, re-suspended in 50 μl and stained with 5 μl MoAb for 30 min at room temperature, washed twice in PBS–2% HPS, and re-suspended in PBS without HPS. FACSort flow cytometer (Becton Dickinson) analyses were performed on 10 000–20 000 cells. Background staining was deducted in calculation of positively staining cells. The percentage of Vβ+ cells was calculated relative to staining with TCRαβ MoAb.
Analysis of the CDR3 of the TCRBV gene
CDR3 analysis was performed as described elsewhere [17]. Briefly, oligo-dT beads (Dynal) were used for extraction of mRNA and cDNA was synthesized using M-MLV Reverse Transcriptase (Gibco) and a BC-specific oligonucleotide primer (C-β-rt: 5′-GCG-CTG-ACG-ATC-TGG-CT-3′). In vitro amplification of TCR genes was performed with beta variable (BV)-specific primers from the T-Cell Receptor Vβ Typing Amplimer Kit (Clontech Labs, Palo Alto, CA) and a FAM5′- or TAMRA5′-labelled BC primer (C-β-5: 5′-CAG-CGA-CCT-CGG-GTG-GGA-3′).
Polymerase chain reaction (PCR) products were analysed using an ABI Prism 377 DNA Sequencer (Perkin Elmer Corp., Foster City, CA). PCR-amplified BV gene segments were diluted 1:10–1:50, mixed with Genescan-500 Rox (Perkin Elmer) and run on a 4.25% denaturing PAGE gel at 24 mA for 2 h. For each BV family, all fragments of equal length migrate for an equal distance on the gel. Using ABI Prism GeneScan Analysis software (Perkin Elmer), the bands produced by migration appear as peaks when plotted and were quantified by peak height. Base pair length was defined by comparison with size standards and converted to corresponding CDR3 amino acid length [17].
Sequencing of individual CDR3 length bands
BV family-specific PCR product and corresponding PHA control material were re-amplified with an internal 33P-labelled BC-specific primer (5′-GAC-CTC-GGG-TGG-GAA-CAC-3′) (T4Polynucleotide Kinase Kit; New England BioLabs, Beverly, MA) and the original BV primers. PCR products were separated on a 6% polyacrylamide gel and the gel transferred to Watman paper and dried before exposure to roentgenographic film. Using the film for alignment, bands were excised from the paper, soaked in distilled water, heated to 95°C for 5 min, and then centrifuged at 7000 g for 5 min. The supernatant was precipitated with 1/10th vol. 3 m sodium acetate and 2.5 vol. 100 m ethanol in the presence of 20 μg glycogen. This precipitate was collected by microcentrifugation, washed in 70% ethanol, dissolved in distilled water, and an aliquot re-amplified using the same BV and BC primers. The resulting PCR was sequenced using a Thermo Sequenase Radiolabeled Terminator Cycle Sequencing Kit (Amersham) and the BC primer with extreme caution to avoid cross-contamination of samples.
Statistical analysis
Student's t-test was used to compare the number of peaks by CDR3 length analysis from all BV families together, between T cell subsets and PHA blast controls. As described elsewhere, peaks in CDR3 length analysis were defined as expanded when significantly higher than expected for that CDR3 length (P < 0.05), based on the CDR3 length distribution in a reference panel of PHA blasts [17]. Student's t-test was used to compare the number of expanded peaks between T cell subsets. The significance of the correspondence in location of expanded peaks between CD8+CD28+ and CD8+CD28− populations from the same donor and BV family was evaluated by a permutation test.
RESULTS
CDR3 length analysis: dominance by relatively few clones within CD8+CD28− T cells
CDR3 length analysis of 22 TCRBV families was performed on CD4+, CD8+CD28+, and CD8+CD28− T cells from six healthy donors. Representative BV CDR3 length analyses are shown in Fig. 1. In 96% of BV families analysed from CD4+ T cells, a number of peaks of almost symmetrically distributed height appear in the CD4+ CDR3 length distributions (Table 1). This pattern is similar to that for the PHA blast cells used as controls, and is reflected in the high number of peaks counted within CD4+ distributions (Table 1). In contrast, in BV analyses of CD8+CD28− T cells, expanded peaks were observed with a frequency of 86%. A polyclonal background, if it exists in this subset, was suppressed to the extent that the average number of peaks (3.4 per BV analysis) was significantly lower than in the other subsets (P < 0.01). CDR3 analyses of CD8+CD28+ T cells were predominantly polyclonal, with an average of 5.9 peaks and a 35% frequency of expanded peaks. When CDR3 analyses were repeated at intervals of up to 2.5 years (donor 1, 20 and 30 months; donor 2, 10 months; donor 3, 3 months), expanded CD8+CD28− peaks were persistent in 84% of cases and CD8+CD28+ peaks in 80%. Expanded peaks within CD4+ analyses were always present upon follow up, but this included only three cases.
Fig. 1.
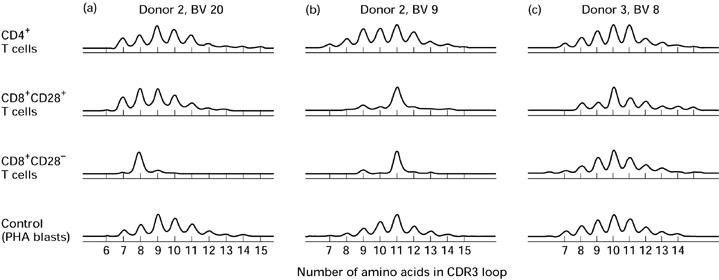
Patterns of beta variable (BV) CDR3 length distribution within T cell subsets. CDR3 length analyses of peripheral blood CD4+, CD8+CD28+ and CD8+CD28− T cells. The ordinates show relative proportion of cellular material sharing the same CDR3 loop length (fluorescence intensity).
Table 1.
Number of total and expanded peaks in beta variable (BV) CDR3 length analyses of T cell subsets

The number of peaks is the mean per CDR3 length analysis, with results from all BV families from all donors grouped together. The s.d. and number of analyses are given in parentheses. The frequency of expanded peaks is the number of expanded peaks detected per BV CDR3 analysis. The number of analyses is given in parentheses. NA, Not applicable.
To examine further the similarity in CDR3 length distribution between the CD8+CD28+ and CD8+CD28− T cell subsets, analyses from the same BV family and donor were paired. Of the expanded CD8+CD28+ peaks, 38% were matched by a peak of the same CDR3 length within CD8+CD28− cells (Fig. 1b). Of the expanded CD8+CD28− peaks, 16% had a matching CD8+CD28+ peak. A permutation test was performed in order to determine whether such a degree of matching could result through chance alone, and this possibility was rejected (P = 0.01).
To determine if peaks were expanded as a result of proliferation of single clones, expanded peaks from each T cell subset were subjected to sequencing. In the vast majority of cases, sequences were dominated by a single clone. This included the one expanded peak chosen from the CD4+ subset, all six peaks from the CD8+CD28+ subset, and 13 of 15 peaks from the CD8+CD28− subset. In one case from the CD8+CD28− subset, two clones were dominant. Three persistent expansions within the CD8+CD28− subset were sequenced, and the presence of the same single dominant clone was confirmed in each case (Fig. 2a).
Fig. 2.
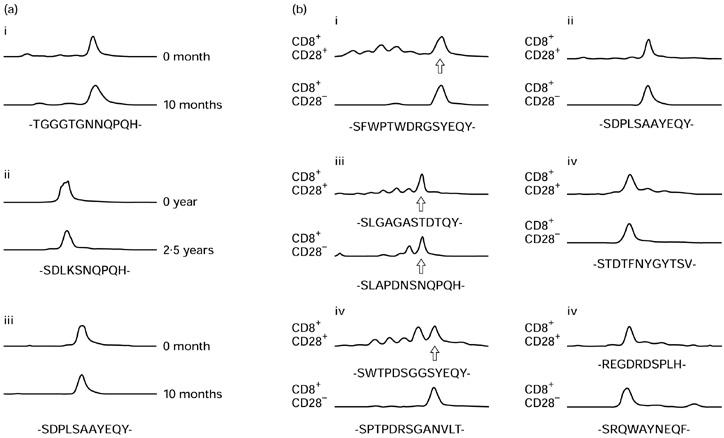
Sequencing of expanded beta variable (BV) CDR3 peaks. (a) Persistence of clones within the CD8+CD28− subset. (b) Expanded peaks of the same length within the CD8+CD28+ and CD8+CD28− subsets. CDR3 amino acid sequences have been deduced from the nucleotide sequences. The abscissae show CDR3 loop length. The ordinates indicate relative proportion of cellular material sharing the same CDR3 loop length (fluorescence intensity). (b) Sequences are reported only once where identical within both CD8+ subsets. In (a) donor 1 is represented in panels i and ii (BV12 and 15), donor 2 in panel iii (BV15). In (b) donor 2 is represented in panels i and ii (BV5.2 and 15), donor 4 in panels iii and iv (BV5.2 and 21), and donor 5 in panels v and vi (BV5.1 and 12).
To determine if identical clonal expansions could be demonstrated within CD8+CD28+ and CD8+CD28− subsets, matching expanded peaks were subjected to sequencing. Results appear in Fig. 2b. Matching expanded peaks were dominated by the same clone in three of six cases. In the other three cases, different dominant clones were found.
Greatly expanded Vβ families amongst CD28− T cells; phenotypic characterization and persistence
The presence of a clonally expanded population of cells, if large enough, will be reflected not only by an altered CDR3 distribution within that BV family, but also by an over-representation of the Vβ family as a whole. In a previous study of healthy subjects, Vβ family representation within peripheral blood lymphocytes was studied using a panel of 12 MoAbs [18]. Three donors with Vβ expansions were investigated further; these persons are the same as donors 1–3 in CDR3 analysis. Donor 1 had Vβ22 CD8+ and Vβ8 CD4+ expansions. Donors 2 and 3 had Vβ5.2 + 3 and Vβ13.3 CD8+ expansions, respectively. By CDR3 analysis and sequencing, these large expansions were dominated by single clones. Co-staining with cell surface markers indicative of state of activation and memory/naive phenotype was performed. Results for the expanded CD8+ Vβ families appear in Fig. 3. These were predominantly CD28− and CD57+, CD11b+, of varying CD45RA/RO phenotype, CD25−, CD69−, CD122−, CD71−, mostly HLA-DR−, and CD95+. Results for the CD4+ Vβ8 family expansion were similar (data not shown). Expansions remained relatively constant for upwards of 4.5 years of observation and, where examined, also appeared to retain their phenotypic characteristics (data not shown). The turnover of cells within the expanded clones was investigated by co-staining with Ki-67, which binds an intracellular antigen expressed in all active stages of the cell cycle [19] (Fig. 4). The percentage of cells staining positive did not appear to be different for lymphocytes belonging to over-represented Vβ families compared with those belonging to all other Vβ families. Similarly, both groups of lymphocytes had a low level of expression of Annexin-V, an early stage marker for apoptosis (data not shown).
Fig. 3.
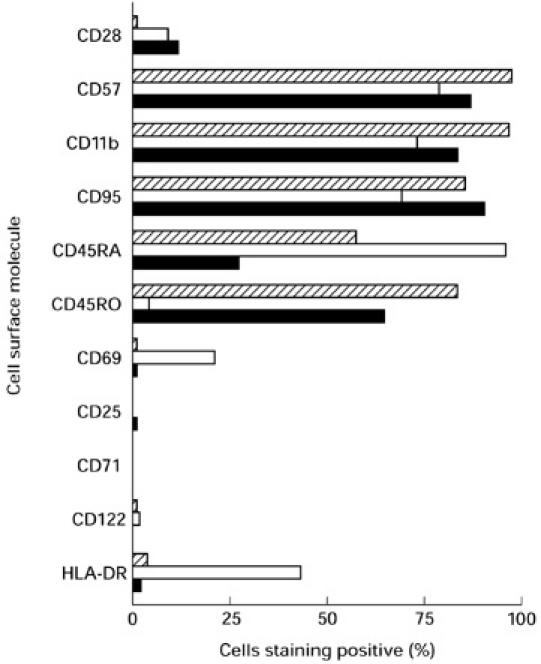
Phenotype of expanded Vβ families. Expression of cell surface molecules within greatly expanded CD8+ T cell Vβ families. Hatched bars, Vβ22 (donor 1); □, Vβ5.2 + 3 (donor 2); ▪, Vβ13.3 (donor 3).
Fig. 4.
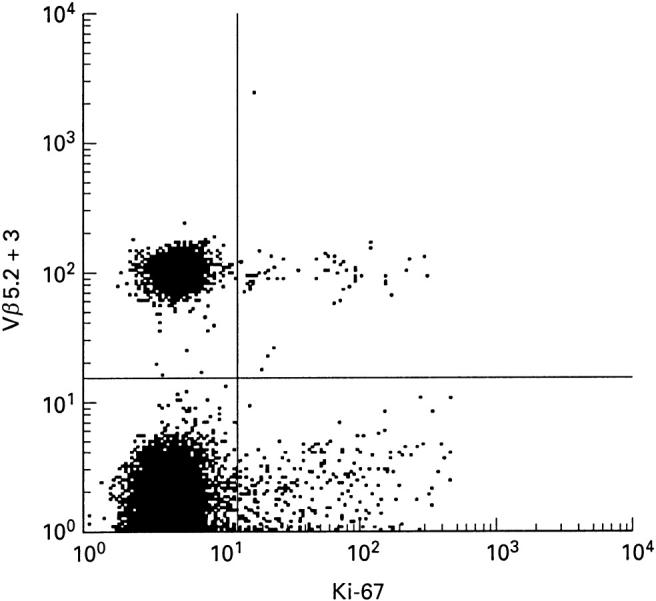
Ki-67 expression on positively selected CD8+ T cells. Comparison of Ki-67 expression within one expanded Vβ family (Vβ5.2 + 3) and all other Vβ families amongst peripheral blood CD8+ T cells from donor 2; 1.0% Ki-67+ of Vβ5.2 + 3+, 1.2% Ki-67+ of Vβ5.2 + 3−. (Similarly, donor 1, 2.7% Ki-67+ of Vβ22+ CD8+ T cells, 1.9% Ki-67+ of Vβ22− CD8+ T cells; 3.3% Ki-67+ of Vβ8+ CD4+ T cells, 1.7% Ki-67+ of Vβ8− CD4+ T cells.)
DISCUSSION
During an immunological event, T cell clones proliferate at the site of antigen presentation. Activated cells are able to travel to other parts of the body via the bloodstream. Most will die by apoptosis, but some will be retained as memory cells. Using TCRBV CDR3 analysis and sequencing, we have studied peripheral blood lymphocytes from healthy donors and looked for evidence of clonal expansions amongst CD4+, CD8+CD28+ and CD8+CD28− T cells. We found that expanded clones are rare within the CD4+ subset, common within the CD8+CD28+ subset, and dominant within the CD8+CD28− subset. Further, most of the clonal expansions detected appeared to persist over many months or even years of observation.
The most likely explanation for the difference in the number of expanded clones between T cell subsets is to be found in the way that antigen is differentially presented to CD4+ and CD8+ T cells. CD8+ T cells respond to antigens present within the antigen-presenting cell itself and presented by MHC class I molecules [20]. Examples of such antigens are tumour antigens and viral antigens. Some of these antigens may persist over months and years, and could explain the observed persistence of expanded CD8+ T cell clones. CD4+ T cells respond to exogenous antigens which are endocytosed by the antigen-presenting cells, processed, and presented by MHC class II molecules [21]. While foreign antigenic material may commonly be present extracellularly during acute infection, release of protein antigens into the extracellular fluid is probably less common for persistent antigens such as those of latent viral infection. Another reason for the more frequent observation of clonal expansions within CD8+ subsets may be that clonal expansion in response to antigen is more vigorous in CD8+ cells than it is in CD4+ cells [22,23]. Third, a dilutional factor may also play some role. Detection of a clone of cells is more difficult if there are more background cells, and normally there are twice as many CD4+ cells as CD8+ cells in the peripheral blood and at least five times as many in the lymphoid tissues [24]. However, the present data show that persistent clonal expansions, although less frequently, occur also within CD4+ T cells. This is consistent with previous reports [9,10,13,14,25,26]. Such expansions have been shown to be more prevalent within the CD28− subset of CD4+ T cells [27,28].
It is now believed that most mature CD8+ T cells express CD28 upon release into the periphery [29–33] and that costimulation via the CD28 molecule is necessary for activation of naive and resting memory CD8+ T cells [34]. Persistently proliferating CD8+CD28+ T cells are believed to lose expression of CD28 after many rounds of cell division [29,30]. Our observation of identical expanded clones within both the CD8+CD28+ and CD8+CD28− subsets supports this notion. If persistent antigen is responsible for the persistently expanded clones observed here, then it may be postulated that after a certain number of cell divisions, the clonally expanded CD8+CD28+ T cells lose CD28 and accumulate as end-stage CD8+CD28− T cell clones. There are, presumably, relatively few persistent antigens, which may explain the observation of a relatively small number of clones within the CD8+CD28− subset. If presentation of antigen is limited to a few cells or to a short period of time, then the release of recently activated, clonally expanded cells into the bloodstream is likely to be of low magnitude, and the proportion of clonally expanded cells in active cell cycle is likely to be small. This is consistent with our observations. A proportion of clonally expanded cells, by definition immunologically experienced cells, also expressed CD45RA, a cell surface marker frequently associated with immunological naivety. As we discuss in another work, however, the CD45RA isoform is a marker for naive cells with low specificity, particularly within the CD8+ T cell subset [33].
Acknowledgments
This study was supported in part by grants from Medinnova, The Sonneborn Charitable Trust, and UNIFOR. Line B. K. Steffarud, Anette M. Syversen, Ingebjørg Knutsen, and Eli Brundtland are acknowledged for technical assistance.
REFERENCES
- 1.Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA. An alpha beta T cell receptor structure at 2.5 A and its orientation in the TCR–MHC complex. Science. 1996;274:209–19. doi: 10.1126/science.274.5285.209. [DOI] [PubMed] [Google Scholar]
- 2.Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384:134–41. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 3.Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. 1993;11:191–212. doi: 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz RH. Models of T cell anergy: is there a common molecular mechanism? J Exp Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz RH. T cell clonal anergy. Curr Opin Immunol. 1997;9:351–7. doi: 10.1016/s0952-7915(97)80081-7. [DOI] [PubMed] [Google Scholar]
- 6.Brinchmann JE, Dobloug JH, Heger BH, Haaheim LL, Sannes M, Egeland T. Expression of costimulatory molecule CD28 on T cells in human immunodeficiency virus type 1 infection: functional and clinical correlations. J Infect Dis. 1994;169:730–8. doi: 10.1093/infdis/169.4.730. [DOI] [PubMed] [Google Scholar]
- 7.Posnett DN, Sinha R, Kabak S, Russo C. Clonal populations of T cells in normal elderly humans: the T cell equivalent to ‘benign monoclonal gammapathy’. J Exp Med. 1994;179:609–18. doi: 10.1084/jem.179.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grunewald J, Jeddi-Tehrani M, Dersimonian H, Andersson R, Wigzell H. A persistent T cell expansion in the peripheral blood of a normal adult male: a new clinical entity? Clin Exp Immunol. 1992;89:279–84. doi: 10.1111/j.1365-2249.1992.tb06945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hingorani R, Choi IH, Akolkar P, Gulwani-Akolkar B, Pergolizzi R, Silver J, Gregersen PK. Clonal predominance of T cell receptors within the CD8+ CD45RO+ subset in normal human subjects. J Immunol. 1993;151:5762–9. [PubMed] [Google Scholar]
- 10.Jeddi-Tehrani M, Grunewald J, Hodara V, Andersson R, Wigzell H. Nonrandom T-cell receptor J beta usage pattern in human CD4+ and CD8+ peripheral T cells. Hum Immunol. 1994;40:93–100. doi: 10.1016/0198-8859(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 11.Fitzgerald JE, Ricalton NS, Meyer AC, West SG, Kaplan H, Behrendt C, Kotzin BL. Analysis of clonal CD8+ T cell expansions in normal individuals and patients with rheumatoid arthritis. J Immunol. 1995;154:3538–47. [PubMed] [Google Scholar]
- 12.Monteiro J, Hingorani R, Choi IH, Silver J, Pergolizzi R, Gregersen PK. Oligoclonality in the human CD8+ T cell repertoire in normal subjects and monozygotic twins: implications for studies of infectious and autoimmune diseases. Mol Med. 1995;1:614–24. [PMC free article] [PubMed] [Google Scholar]
- 13.Grunewald J, Wigzell H. T-cell expansions in healthy individuals. Immunologist. 1996;4:99–103. [Google Scholar]
- 14.Shen DF, Doukhan L, Kalams S, Delwart E. High-resolution analysis of T-cell receptor beta-chain repertoires using DNA heteroduplex tracking: generally stable, clonal CD8+ expansions in all healthy young adults. J Immunol Methods. 1998;215:113–21. doi: 10.1016/s0022-1759(98)00066-0. [DOI] [PubMed] [Google Scholar]
- 15.Walker BD, Plata F. Cytotoxic T lymphocytes against HIV. AIDS. 1990;4:177–84. doi: 10.1097/00002030-199003000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Funderud S, Nustad K, Lea T, Vartdal F, Gaudernack G, Stenstad P, Ugelstad J. Fractionation of lymphocytes by immunomagnetic beads. In: Klaus GB, editor. Lymphocytes: a practical approach. Oxford: IRL Press; 1987. pp. 55–65. [Google Scholar]
- 17.Mugnaini EN, Egeland T, Syversen AM, Spurkland A, Brinchmann JE. Molecular analysis of the complementarity determining region 3 of the human T cell receptor β chain. Establishment of a reference panel of CDR3 lengths from phytohaemagglutinin activated lymphocytes. J Immunol Methods. 1999;223:207–16. doi: 10.1016/s0022-1759(99)00004-6. [DOI] [PubMed] [Google Scholar]
- 18.Brinchmann JE, Janson CH, Klem LB, Haaheim LL, Spurkland A. Strict adherence to a common rank order of T-cell receptor V beta usage in human leucocyte antigen disparate individuals. Scand J Immunol. 1996;44:179–84. doi: 10.1046/j.1365-3083.1996.d01-294.x. [DOI] [PubMed] [Google Scholar]
- 19.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–5. [PubMed] [Google Scholar]
- 20.Zinkernagel RM, Doherty PC. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature. 1974;248:701–2. doi: 10.1038/248701a0. [DOI] [PubMed] [Google Scholar]
- 21.Rosenthal AS, Shevach EM. Function of macrophages in antigen recognition by guinea pig T lymphocytes. I. Requirement for histocompatible macrophages and lymphocytes. J Exp Med. 1973;138:1194–212. doi: 10.1084/jem.138.5.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchmeier MJ, Welsh RM, Dutko FJ, Oldstone MB. The virology and immunobiology of lymphocytic choriomeningitis virus infection. Adv Immunol. 1980;30:275–331. doi: 10.1016/s0065-2776(08)60197-2. [DOI] [PubMed] [Google Scholar]
- 23.Callan MF, Steven N, Krausa P, et al. Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat Med. 1996;2:906–11. doi: 10.1038/nm0896-906. [DOI] [PubMed] [Google Scholar]
- 24.Røsok BI, Bostad L, Voltersvik P, Bjerknes R, Olofsson J, Åsjö B, Brinchmann JE. Reduced CD4 cell counts in blood do not reflect CD4 cell depletion in tonsillar tissue in asymptomatic HIV-1 infection. AIDS. 1996;10:F35–38. [PubMed] [Google Scholar]
- 25.Waase I, Kayser C, Carlson PJ, Goronzy JJ, Weyand CM. Oligoclonal T cell proliferation in patients with rheumatoid arthritis and their unaffected siblings. Arthritis Rheum. 1996;39:904–13. doi: 10.1002/art.1780390606. [DOI] [PubMed] [Google Scholar]
- 26.Walser-Kuntz DR, Weyand CM, Goronzy JJ. Influence of antigenic experience on BJ gene segment usage in human CD4+ T cells. Int Immunol. 1997;9:1785–92. doi: 10.1093/intimm/9.12.1785. [DOI] [PubMed] [Google Scholar]
- 27.Weyand CM, Brandes JC, Schmidt D, Fulbright JW, Goronzy JJ. Functional properties of CD4+CD28- T cells in the aging immune system. Mech Ageing Dev. 1998;102:131–47. doi: 10.1016/s0047-6374(97)00161-9. [DOI] [PubMed] [Google Scholar]
- 28.Schirmer M, Vallejo AN, Weyand CM, Goronzy JJ. Resistance to apoptosis and elevated expression of bcl-2 in clonally expanded CD4+CD28- T cells from rheumatoid arthritis patients. J Immunol. 1998;161:1018–25. [PubMed] [Google Scholar]
- 29.Monteiro J, Batliwalla F, Ostrer H, Gregersen PK. Shortened telomeres in clonally expanded CD28–CD8+ T cells imply a replicative history that is distinct from their CD28+CD8+ counterparts. J Immunol. 1996;156:3587–90. [PubMed] [Google Scholar]
- 30.Effros RB, Allsopp R, Chiu CP, et al. Shortened telomeres in the expanded CD28–CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS. 1996;10:F17–22. doi: 10.1097/00002030-199607000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, van Lier RA. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. 1997;186:1407–18. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mugnaini EN, Spurkland A, Egeland T, Sannes M, Brinchmann JE. Demonstration of identical expanded clones within both CD8+CD28+ and CD8+CD28- T cell subsets in HIV type 1-infected individuals. Eur J Immunol. 1998;28:1738–42. doi: 10.1002/(sici)1521-4141(199805)28:05<1738::aid-immu1738>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 33.Mugnaini EN, Haaheim LL, Sannes M, Brinchmann JE. In vivo expansion coincident with excessive in vitro cell death within the memory subset of CD8+ T-cells in HIV-1 infection. AIDS Res Hum Retrovir. 1999;15:265–72. doi: 10.1089/088922299311448. [DOI] [PubMed] [Google Scholar]
- 34.Azuma M, Phillips JH, Lanier LL. CD28- T lymphocytes. Antigenic and functional properties. J Immunol. 1993;150:1147–59. [PubMed] [Google Scholar]