

Abstract
IL-12 modulates Th1 immune response during chronic colitis. Mechanisms regulating IL-12 synthesis in human intestine are poorly understood. The aim of this study was to investigate the effect of IFN-γ and PGE2 on lipopolysaccharide (LPS)-stimulated LPMC IL-12 production. Normal LPMC cultures were run in the presence or absence of IFN-γ and/or PGE2 before LPS stimulation. To examine the role of endogenous PGE2 on LPS-stimulated IL-12 release, LPMC cultures were added of indomethacin before LPS stimulation. IL-12, IL-10 and IL-8 were measured by ELISA. No IL-12 was detected in either unstimulated or LPS-stimulated LPMC cultures. In contrast, LPMC released IL-8 (650 ± 125 pg/ml) and IL-10 (75 ± 25 pg/ml) in response to LPS. Treatment of LPMC with IFN-γ facilitated LPS-stimulated IL-12, whereas it completely abrogated IL-10 production. IL-12 release by LPMC stimulated with IFN-γ and LPS was significantly inhibited by exogenous IL-10. The addition of PGE2 to IFN-γ-treated LPMC cultures inhibited in a dose-dependent manner LPS-induced IL-12 secretion. Furthermore, IL-12 was detectable (85 ± 25 pg/ml) in the supernatants of LPMC cultures treated with indomethacin and LPS. In contrast to the effect on IL-12, PGE2 significantly augmented LPS-stimulated LPMC IL-10 production. However, the inhibition of IL-12 by PGE2 was only partially reversed by anti-IL-10. In a simplified model of LPS tolerance, we finally showed that monocyte-derived macrophages exhibited reduced IL-12 production after repeat LPS stimulation. In these cell cultures, indomethacin abrogated the induction of LPS desensitization. IFN-γ and PGE2 modulate differently the LPMC responsiveness to LPS in terms of IL-12 synthesis.
Keywords: IL-12, interferon-gamma, lipopolysaccharide, prostaglandin-E2, intestinal inflammation
INTRODUCTION
IL-12, a heterodimeric cytokine composed of two subunits (p35 and p40), is produced by activated antigen-presenting cells (APC) mainly in response to bacteria or bacterial products [1–3]. IL-12 plays a pivotal role in generating Th1-mediated inflammatory responses by priming T and natural killer (NK) cells for high IFN-γ production [1–3]. IFN-γ enhances in turn the ability of APC to produce IL-12, triggering a positive feedback mechanism capable of amplifying APC activation and maintaining a Th1-type immune response [1,2,4]. In murine models, IFN-γ proved to be necessary for inducing macrophage IL-12 production, supporting the hypothesis that during some Th1-mediated diseases IL-12 may not be the first cytokine generated [5]. It is, however, widely shown that the synthesis of IL-12 by APC in response to bacterial stimulation is also regulated by cytokines/factors with deactivating effects on the producer cells [1–3].
IL-12 is expressed and actively released by intestinal LPMC in Crohn's disease (CD) [6,7], a chronic inflammatory process involving the alimentary tract and characterized by Th1-mediated phenomena [8–11]. Evidence has also accumulated to indicate that IL-12 modulates the differentiation of Th1-type lymphocytes during intestinal inflammation [7,12,13].
Mechanisms regulating IL-12 production in the human intestinal mucosa are not understood. Studies from experimental models suggest that the development of Th1-type cytokine-mediated chronic colitis requires the presence of non-pathogenic bacterial flora and that the inflammation may be driven by IL-12 [14,15]. Lipopolysaccharide (LPS), a bacterial product largely present in the gut lumen, is a powerful inducer of IL-12 in vitro [1–4]. It has been shown, however, that the capability of releasing IL-12 in response to LPS is impaired in macrophages isolated from the normal gut mucosa [6]. In addition, IL-12 has been hardly detected in normal intestinal mucosa [6,7]. Taken together, these observations suggest that in normal intestine IL-12 production is a tightly regulated function and that local factors may operate in down-regulating IL-12.
PGE2 is an important mediator of the acute inflammatory response. At the site of tissue injury PGE2 induces erythema, oedema, and hyperalgesia [16,17]. PGE2 is also known to inhibit B and T lymphocyte proliferation, T lymphocyte IL-2 and IFN-γ synthesis, and IL-2 receptor expression [18]. Furthermore, in the presence of bacterial products, such as LPS, PGE2 acts as an inhibitor of APC functions [18–21]. Consistent with these observations, PGE2 is normally synthesized in human intestine where it seems to be determinant in preventing APC-derived cytokine secretion [22,23]. We therefore, hypothesized that in the human intestinal mucosa PGE2 contributes to down-regulate IL-12 production, counterbalancing the effects of IFN-γ.
In the present study, we report data indicating that IFN-γ and PGE2 modulate differently the responsiveness of normal LPMC to LPS in terms of IL-12 production.
MATERIALS AND METHODS
Mucosal samples and mononuclear cell isolation
Mucosal samples were obtained from the macroscopically and microscopically unaffected areas of 16 surgical colon cancer specimens. Autologous peripheral blood mononuclear cells (PBMC) were obtained from nine patients. PBMC from six healthy subjects were also available. The study was approved by the local Department Ethical Committee. LPMC were isolated by the DTT–EDTA–collagenase sequence as previously described [6,9]. The isolated cells were counted and checked for viability using 0.1% trypan blue (viability ranged from 91% to 95%). PBMC were isolated by density gradient centrifugation (Lymphoprep; Nycomed Pharma, Oslo, Norway). When used, purified macrophages and monocytes were obtained by seeding LPMC or PBMC, respectively (3 × 106/ml), in microtitre plates and on chamber slides (Lab-Tek; Nunc, Roskilde, Denmark) coated with 0.1% (w/v) gelatin (Sigma Chemical Co., St Louis, MO). After 3 h at 37°C, non-adherent cells were removed by washings with prewarmed PBS 1× (Sigma), while adherent cells were collected by gentle washings with cold PBS 1×. Adherent cell preparations contained consistently < 5% contaminating CD3+ cells, as assessed by FACS analysis (data not shown). In order to control monocyte/macrophage morphology in adherence-separated LPMC or PBMC cultures, adherent cells on chamber slides were fixed in ethanol and stained by haematoxylin.
Cell cultures
Both total and adherence-separated cells were cultured in complete medium consisting of RPMI 1640 supplemented with 10% fetal calf serum (FCS), 1% l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (all Sigma). Both LPMC and PBMC (concentration 3 × 106 cells/ml) cultures were run with or without the initial addition of LPS (Escherichia coli) (1 μg/ml) or Staphylococcal enterotoxin B (SEB; 1 μg/ml) (both Sigma) for 24 h. Parallel experiments were performed using adherent cells (at a concentration of 5 × 105 cells/ml). After 24 h of culture, cell-free supernatants were collected and stored at −80°C until tested. LPMC and PBMC were also treated with graded doses of recombinant human IFN-γ (Becton Dickinson Labware, Bedford, MA) (final concentration ranging from 1 ng/ml to 250 ng/ml) or recombinant human IL-4 (Sigma) (final concentration ranging from 1 ng/ml to 250 ng/ml) for 32 h before LPS stimulation (1 μg/ml) for a further 24 h. Parallel cultures were added of an IFN-γ antiserum (1:100 final dilution; kindly supplied by Dr M. R. Capobianchi, Istituto di Virologia, Università La Sapienza, Roma, Italy) or a neutralizing IL-4 antibody (1:100 final dilution; R&D Systems, Minneapolis, MN). Experiments were also performed by stimulating LPMC with IFN-γ (100 ng/ml final concentration) and recombinant human IL-10 (10 ng/ml final concentration; Sigma). At the end of the culture period, cell-free supernatants were collected and stored at −80°C until tested.
To examine the effect of PGE2 on IL-12 production, LPMC were treated with IFN-γ (100 ng/ml) in the presence or absence of PGE2 (final concentration ranging from 10−5 m to 10−8m; provided by Dr G. De Sarro, University of Catanzaro) for 32 h before LPS (1 μg/ml) stimulation for a further 24 h. Cultures were also treated with PGE2 alone before LPS stimulation. To prove further that endogenous PGE2 modulates LPS-stimulated IL-12 production, LPMC cultures were provided with 100 μg/ml indomethacin for 32 h before LPS (1 μg/ml) stimulation. In order to explore the possibility that PGE2 modulates IL-12 release through the induction of IL-10, LPMC cultures treated with IFN-γ (100 ng/ml) and PGE2 (10−5m) were added of a neutralizing human IL-10 MoAb (100 ng/ml final concentration) (clone 23738.11; Sigma). At the end of the culture period, cell-free supernatants were collected and stored at −80°C until tested.
IL-12 production by monocyte-derived macrophages pretreated with low doses of LPS
To investigate whether monocyte-derived macrophages pretreated with low concentrations of LPS produce IL-12 upon restimulation with LPS, adherent mononuclear cells from peripheral blood were resuspended in complete medium at a concentration of 1 × 106 cells/ml and cultured for 7 days. Thereafter, cells were washed and cultured again in complete medium in the presence or absence of LPS (200 pg/ml final concentration) or LPS plus indomethacin (100 μg/ml final concentration) for 2 days (primary culture). At the end of the culture period, cells were extensively washed and stimulated again with medium alone or 100 ng/ml LPS for a further 24 h (secondary culture). Thereafter, cell-free supernatants were collected and stored at −80°C until tested.
Cytokine assay
IL-12, IL-10 and IL-8 were measured in the cell culture supernatants using sensitive ELISA kits (Amersham Int. plc, Slough, UK). The minimum detectable concentration was 5 pg/ml for both IL-12 and IL-10, and 10 pg/ml for IL-8. IL-12 was measured using a commercially available kit that detects both IL-12 p40 and the bioactive IL-12 p70 heterodimer.
Statistical analysis
Student's t-test was used for statistical analysis of the data.
RESULTS
Normal LPMC fail to release IL-12 after LPS stimulation
No IL-12 was detected in the supernatants of total or adherence-separated LPMC cultures incubated with medium alone. Similarly, LPMC failed to release IL-12 after LPS stimulation. In contrast, IL-12 was measured in the supernatants of both PBMC (160 ± 10 pg/ml) and monocyte-derived macrophage (290 ± 40 pg/ml) cultures provided with LPS. Both total and adherence-separated LPMC were, however, able to release IL-12 in response to SEB (38 ± 8.5 pg/ml and 25 ± 4.0 pg/ml, respectively). Importantly, the inability of LPS-stimulated LPMC to produce IL-12 was not the result of a global reduction in protein synthesis, because these cells released IL-8 (650 ± 125 pg/ml versus 120 ± 30 pg/ml in unstimulated LPMC; P < 0.001). Furthermore, LPMC viability in the presence of LPS was > 90% as assessed by trypan blue exclusion.
IFN-γ facilitates IL-12 release in LPS-stimulated LPMC cultures
IFN-γ is a major activator of monocytes/macrophages [24]. There is now evidence indicating that IFN-γ specifically influences monocyte IL-12 production [4,25]. In addition, studies from human and animal models indicate that IFN-γ promotes cytokine secretion by endotoxin-resistant cells [26,27]. To examine whether IFN-γ could restore IL-12 production by normal intestinal mononuclear cells, LPMC were preincubated with graded doses of IFN-γ before LPS stimulation. IFN-γ alone did not induce IL-12 production. IFN-γ, however, promoted, in a dose-dependent fashion, IL-12 release by LPS-stimulated LPMC (Table 1 and Fig. 1). In both total and adherence-separated LPMC cultures, the amount of IL-12 produced in response to LPS after treatment with 100 ng/ml IFN-γ (320 ± 70 pg/ml and 250 ± 20 pg/ml, respectively) was two and four times higher than that measured in cell cultures provided with 25 ng/ml or 10 ng/ml IFN-γ, respectively (180 ± 35 pg/ml and 140 ± 30 pg/ml in 25 ng/ml IFN-γ-stimulated cells; 80 ± 15 pg/ml and 60 ± 10 pg/ml in 10 ng/ml IFN-γ-stimulated cells) (Fig. 1). The addition of an IFN-γ antiserum decreased by nearly 10 times the amount of IL-12 released by total LPMC treated with 100 ng/ml IFN-γ (320 ± 70 pg/ml versus 45 ± 18 pg/ml; P < 0.0001), and completely abolished that measured in cell cultures treated with 25 ng/ml or 10 ng/ml IFN-γ. No inhibitory effect was seen with a non-relevant control antibody (anti-IL-4).
Table 1.
IL-12 release by LPMC or peripheral blood mononuclear cells (PBMC)

Cells were cultured and preincubated in the presence of medium or IL-4 (100 ng/ml final concentration) or IFN-γ (100 ng/ml final concentration) for 32 h before stimulating with complete medium or Staphylococcal enterotoxin B (SEB; 1 μg/ml final concentration) or lipopolysaccharide (LPS; 1 μg/ml final concentration) for a further 24 h. Results are mean ± s.d. of six experiments. IL-12 values are expressed as pg/ml. Und., Undetectable.
Fig. 1.
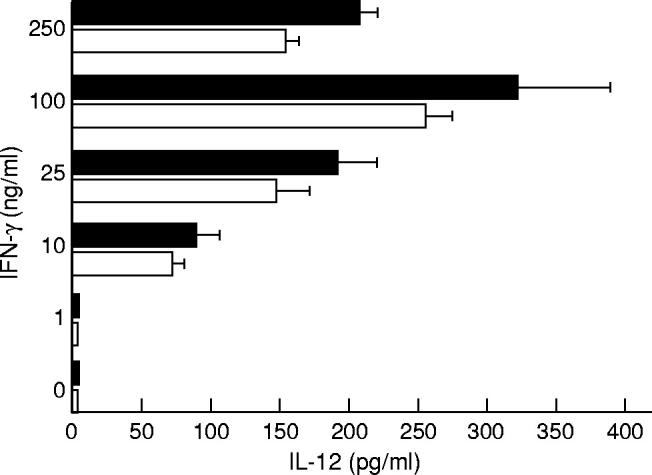
IFN-γ promotes IL-12 release by both total (▪) and adherence-separated LPMC (□) after exposure to lipopolysaccharide (LPS). Cells were incubated with graded doses of IFN-γ or medium for 32 h before stimulation with 1 μg/ml LPS for a further 24 h. IL-12 was measured by ELISA. Data are the mean of all experiments; bars indicate 1 s.d.
The ability of phagocytes to produce IL-12 is regulated by several cytokines with activating or deactivating effects on the producer cells. IL-10 has been described as a strong inhibitor of monocyte IL-12 synthesis [28]. Since IFN-γ is capable of affecting IL-10 production in monocytic cell lines [29], we hypothesized that IFN-γ could modulate LPS-stimulated LPMC IL-12 synthesis through the inhibition of endogenous IL-10. To address this issue, we first measured IL-10 in the culture supernatants of LPMC treated with 100 ng/ml IFN-γ before LPS stimulation. IFN-γ completely inhibited LPS-stimulated LPMC IL-10 secretion. To prove further that the inhibition of endogenous IL-10 may be a mechanism by which IFN-γ promotes IL-12 synthesis, we treated LPMC with IFN-γ (100 ng/ml) in the presence or absence of recombinant human IL-10 (10 ng/ml final concentration) for 32 h before LPS stimulation. Exogenous IL-10 significantly decreased the release of IL-12 by IFN-γ-treated LPMC after LPS stimulation. The inhibitory effect of IL-10 was completely abolished by the addition of a neutralizing IL-10 antibody (100 ng/ml final concentration) (Table 2).
Table 2.
Effect of recombinant human IL-10 on IL-12 release by LPMC

LPMC were cultured and preincubated with medium alone or IFN-γ (100 ng/ml final concentration) in the presence or absence of rhIL-10 (10 ng/ml final concentration) or rhIL-10 and anti-IL-10 (100 ng/ml) for 32 h before lipopolysaccharide (LPS) stimulation. IL-12 was measured by ELISA. Results are indicated as mean ± s.d. IL-12 values are expressed as pg/ml. Und., Undetectable. Pre-incubation with IL-10 significantly decreased IL-12 secretion by IFN-γ-treated LPMC after LPS exposure (P < 0.01). The effect of IL-10 was completely inhibited by anti-IL-10.
IL-4, originally described as a factor secreted by type 2 cells, can prime peripheral monocytes for optimal IL-12 production in response to LPS stimulation [28]. To investigate whether IL-4 facilitated IL-12 synthesis by LPS-stimulated LPMC, cell cultures were added of IL-4 for 32 h before LPS stimulation. No IL-12 was measured in the culture supernatants of either total or adherence-separated LPMC preincubated with IL-4 and then exposed to LPS. In contrast, IL-4 significantly enhanced SEB-stimulated LPMC IL-12 production (Table 1) (P < 0.001). In addition, IL-4 induced a significant increase in the amount of IL-12 released by LPS-stimulated monocytes (Table 1) (P < 0.0001).
PGE2 mediates LPMC hyporesponsiveness to LPS
PGE2, in the presence of bacterial products, acts as a powerful inhibitor of APC cytokine production [18–23]. In order to investigate whether PGE2 modulates IL-12 production in human intestine, LPMC were preincubated with graded doses of PGE2 and 100 ng/ml IFN-γ for 32 h before LPS stimulation.
As shown in Fig. 2, PGE2 inhibited, in a dose-dependent manner, the production of IL-12, yielding complete inhibition at 10−5 m PGE2. As LPS stimulates the induction of PGE2 [18], in selected experiments indomethacin was added to LPMC cultures to examine LPS-stimulated IL-12 production in the absence of de novo PGE2 synthesis. No IL-12 was detected in the supernatants of LPMC cultures provided with indomethacin alone. In contrast, pre-exposure of LPMC to indomethacin facilitated IL-12 release in response to LPS (85 ± 25 pg/ml). The addition of PGE2 (10−5 m final concentration) to the LPMC cultures provided with indomethacin resulted in complete inhibition of LPS-stimulated LPMC IL-12 release. Taken together, these data indicate that PGE2 is a powerful inhibitor of IL-12 production in human LPMC. Our next goal was to analyse potential mechanisms by which PGE2 down-regulates LPS-stimulated LPMC IL-12 production. Since it is known that PGE2 enhances IL-10 production in human PBMC [19], we investigated the possibility that the inhibitory effect of PGE2 was mediated through the induction of IL-10. Treatment of LPMC with PGE2 significantly augmented LPS-stimulated IL-10 release (265 ± 15, 180 ± 20 and 135 ± 22 pg/ml after 10−5 m, 10−6 m, and 10−7 m PGE2,respectively, versus 74 ± 25 pg/ml) (P < 0.03) (Fig. 3). In agreement with these results, treatment of LPMC with indomethacin resulted in a reduced secretion of IL-10 after LPS stimulation (18 ± 5 pg/ml) (P = 0.03). Further experiments were therefore performed to establish the role of endogenous IL-10 in PGE2-induced inhibition of IL-12 production. LPMC treated with 100 ng/ml IFN-γ were cultured in the presence or absence of PGE2(10−5m) and anti-IL-10 (100 ng/ml) before LPS stimulation. As shown in Fig. 4, PGE2-induced inhibition of IL-12 release in IFN-γ-treated LPMC cultures was only partially reversed by anti-IL-10. This result was not dependent on possible efficacy problems of the anti-IL-10, because this antibody was capable of completely blocking the effect of rhIL-10 on IL-12 secretion in LPMC cultures preincubated with IFN-γ and stimulated with LPS (Table 2).
Fig. 2.
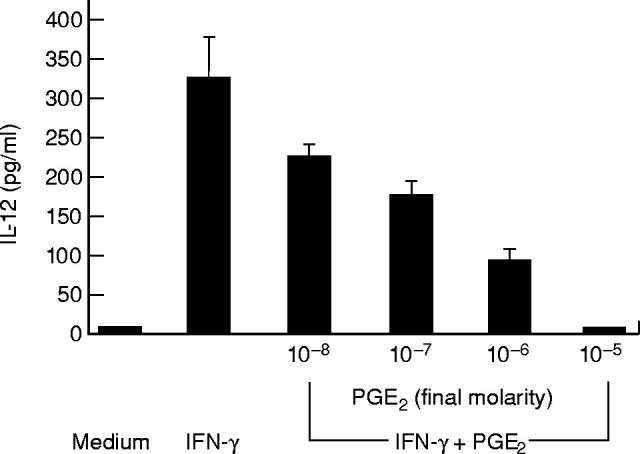
Effect of PGE2 on IL-12 release by IFN-γ-stimulated LPMC exposed to lipopolysaccharide (LPS). Cells were stimulated with medium or 100 ng/ml IFN-γ in the presence or absence of graded doses of PGE2 for 32 h before treating 1 μg/ml LPS for a further 24 h. IL-12 was measured by ELISA. Data are the mean of all experiments; vertical bars indicate 1 s.d.
Fig. 3.
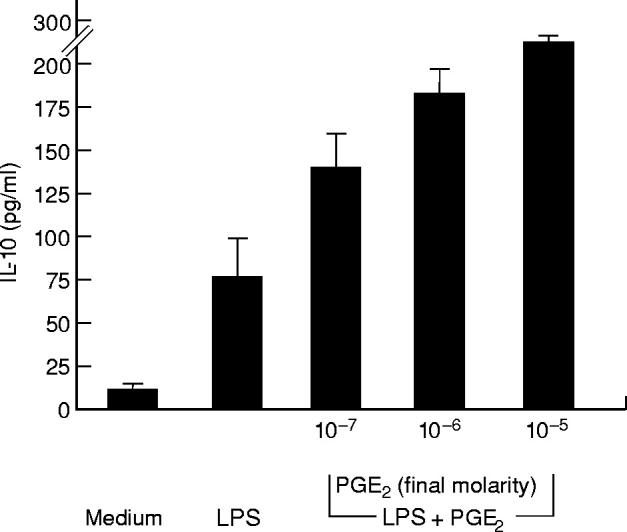
Effect of PGE2 on IL-10 release by LPMC exposed to lipopolysaccharide (LPS). Cells were stimulated with nothing or LPS (1 μg/ml) for 24 h. In parallel experiments LPMC were treated with LPS and graded doses of PGE2 for 24 h. IL-10 was measured by ELISA. Data are the mean of all experiments; vertical bars indicate 1 s.d.
Fig. 4.
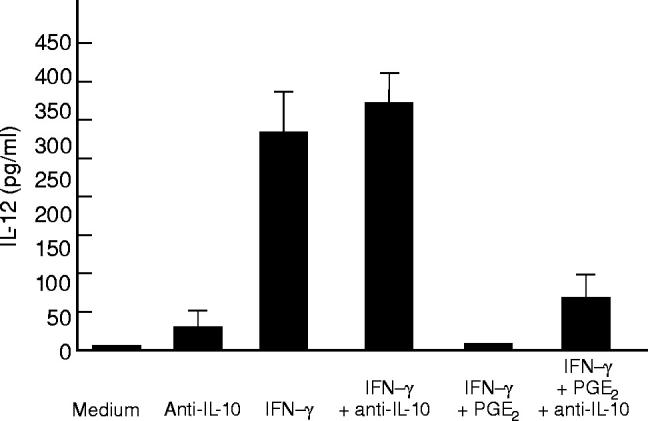
Effect of anti-IL-10 on IL-12 release by LPMC stimulated with lipopolysaccharide (LPS). Cells were treated with medium or 100 ng/ml IFN-γ in the presence or absence of PGE2 (10−5 m) for 32 h (first stimulation) before treating with 1 μg/ml LPS for a further 24 h (second stimulation). Parallel cultures were added of an anti-IL-10 (100 ng/ml final concentration) at the time of the first stimulation. IL-12 was measured by ELISA. Data are the mean of all experiments; vertical bars indicate 1 s.d.
Human monocyte-derived macrophages exhibit reduced IL-12 production after repeat LPS stimulation
Although monocytes/macrophages respond to primary encounter with LPS by releasing cytokines, repeat LPS exposure renders these cells refractory to a further challenge with LPS [26,30]. This phenomenon, termed LPS tolerance or desensitization, may be reproduced in vitro by using different types of APC [26,30]. During the isolation procedure LPMC may be exposed to substantial concentrations of LPS. LPMC might therefore be desensitized to further LPS stimulation. To examine whether IL-12 production by LPS-stimulated APC can be affected by pre-exposure to LPS, monocyte-derived macrophages were treated with low concentrations of LPS for 2 days, washed and stimulated again with 100 ng/ml LPS for a further 24 h. No IL-12 was measured in the supernatants of unstimulated monocyte-derived macrophages. Macrophages did respond to the primary LPS exposure by releasing IL-12 (100 ± 15 pg/ml and 270 ± 42 pg/ml after stimulation with 200 pg/ml and 100 ng/ml LPS, respectively). However, macrophages cultured in the presence of low-dose LPS exhibited a decreased IL-12 release after a secondary challenge with LPS (Table 3). No inhibition of IL-8 release was observed in LPS-desensitized macrophage cultures (Table 3).
Table 3.
IL-12 and IL-8 production by control and lipopolysaccharide (LPS)-primed monocyte-derived macrophages

Human monocyte-derived macrophages were cultured in complete medium at a concentration of 1 × 106/ml in the presence or absence of 200 pg/ml LPS and/or indomethacin for 2 days (primary culture). At the end of the culture period, cells were extensively washed and treated with medium or LPS (100 ng/ml) for a further 24 h (secondary culture). IL-12 and IL-8 release was measured by ELISA and their values are expressed as pg/ml. Und, Undetectable. Results are mean ± s.d. of six experiments.
PGE2 is a major product of LPS stimulation [18]. We therefore explored the possibility that PGE2 could inhibit IL-12 production in LPS-tolerant macrophage cultures. To examine this issue, macrophages were treated with low-dose LPS in the presence or absence of indomethacin for 32 h, then washed and stimulated again with 100 ng/ml LPS and/or medium for 24 h. Macrophages cultured in the presence of indomethacin maintained the capability of releasing IL-12 in response to a secondary stimulation with LPS (180 ± 25 pg/ml) (Table 3).
DISCUSSION
Our study confirms and expands the concept that molecules released during inflammation may influence IL-12 production by LPS-stimulated APC. We here provide evidence that IFN-γ and PGE2 have opposing effects on IL-12 production by LPS-stimulated LPMC.
No IL-12 was measured in LPMC cultures provided with IFN-γ alone, confirming the observation that IFN-γ is not sufficient to completely activate IL-12 genes [4,25]. However, in both total and adherence-separated LPMC exposed to LPS, IFN-γ evoked IL-12 release in a dose-dependent manner. The effect of IFN-γ on IL-12 release appeared to be specific because it was inhibited by an IFN-γ antiserum. These findings are supported by the observations that IFN-γ is capable of promoting the synthesis of other cytokines in LPS-desensitized cell cultures and that endotoxin-resistant macrophages bearing the lpsd mutation respond to IFN-γ in terms of cytokine secretion [26,27]. Our data are moreover consistent with studies showing that, in other cell systems, IFN-γ may specifically enhance LPS-induced IL-12 secretion [1–4,25]. Since IFN-γ is capable of affecting the intestinal permeability and facilitating the influx of bacterial products from the lumen [31,32], these results suggest that IFN-γ can play an important role in inducing IL-12 within the gut mucosa. In contrast to the effect on IL-12, IFN-γ completely inhibited IL-10 secretion by LPS-stimulated LPMC. When recombinant IL-10 was added to IFN-γ-treated cultures, IL-12 release by LPS-stimulated LPMC was remarkably decreased, suggesting that inhibition of endogenous IL-10 may be a mechanism by which IFN-γ modulates LPS-stimulated LPMC IL-12 production.
At the intestinal mucosal level, where luminal (dietary, bacterial and viral) antigens are continuously interacting with immune cells, multiple and complex mechanisms operate in promoting local tolerance. Particularly, counter-balancing molecules seem to be determinant in promoting or blocking the occurrence of chronic mucosal inflammation [33,34]. PGE2, normally synthesized by human intestinal cells, has been shown tomodulate selectively both humoral and cellular response at multiple levels [18]. Particularly, PGE2 and similar cAMP-elevating agents inhibit Th1-type cytokines [35,36] and IL-12 production induced by LPS [19]. Furthermore, PGE2 seems to play a role in inducing tolerance in vivo [21]. In agreement with these observations, we found that PGE2 in a dose-dependent manner inhibited IL-12 release by LPMC stimulated with IFN-γ and LPS. As indomethacin, a powerful inhibitor of PGE2 synthesis, facilitated IL-12 release by LPS-stimulated LPMC, it is plausible that endogenous PGE2 acts in preventing IL-12 synthesis in human intestine. The ability of locally released PGE2 to suppress intestinal macrophage cytokine production has been previously reported. Rugtveit et al. showed that LPMC tumour necrosis factor-alpha (TNF-α) release was enhanced by indomethacin [22], whereas Nathens et al. suggested that, after exposure to bacterial products, endogenous PGE2 down-regulates TNF-α secretion in co-cultures of intestinal epithelial and mononuclear cells [23]. Taken together, these observations suggest that PGE2 may alter the activation state of intestinal macrophages, making them refractory to microbial stimulation. This hypothesis is also supported by in vivo studies showing that indomethacin and other cyclooxygenase inhibitors may contribute to the abnormal activation of the immune system and promote specific inflammatory enteropathies in the presence of luminal bacteria [37,38].
Consistent with other studies [19], we also showed that PGE2 augmented in a dose-dependent fashion IL-10 secretion by LPS-stimulated LPMC. Together with the demonstration that IL-10 is a strong inhibitor of IL-12 production, these findings prompted us to test the hypothesis that PGE2 could modulate LPS-stimulated LPMC IL-12 synthesis through the induction of IL-10. However, inhibition of IL-12 production by PGE2 was only partially reversed by anti-IL-10, suggesting that the induction of IL-10 is not the main mechanism by which PGE2 modulates LPMC IL-12 production.
Repeat LPS stimulation renders cells refractory to further LPS challenges. This phenomenon may be successfully reproduced in vitro [26,30]. We here report data indicating that human monocyte-derived macrophages treated with low concentrations of LPS release scarce amounts of IL-12 upon restimulation with LPS. This phenomenon does not seem to be due to a state of complete macrophage deactivation, because the same cells were capable of producing IL-8 in response to LPS. The capacity of indomethacin to restore IL-12 production by macrophages cultured in the presence of low concentrations of LPS strongly supports the role of PGE2 in promoting the LPS tolerance phenomenon. Interestingly, while this manuscript was in preparation, it was reported that LPS-desensitized dendritic cells are poor sources of IL-12 and that PGE2 may replace LPS for the induction of LPS tolerance [39]. As intestinal macrophages may be exposed to trace amounts of bacterial products either during the isolation procedure or in vivo, it is therefore conceivable that the defective LPMC IL-12 production in response to LPS may be due to a LPS tolerance phenomenon.
In conclusion, our study shows that IFN-γ or PGE2 regulate differently LPMC IL-12 production in response to LPS.
REFERENCES
- 1.Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. 1995;13:251–76. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 2.Trinchieri G. Interleukin-12: a cytokine produced by antigen-presenting cells with immunoregulatory functions in the generation of T-helper cells type 1 and cytotoxic lymphocytes. Blood. 1994;84:4006–27. [PubMed] [Google Scholar]
- 3.Hendrzak JA, Brunda MJ. Biology of disease. Interleukin-12. Biology activity, therapeutic utility, and role in disease. Lab Invest. 1995;72:619–37. [PubMed] [Google Scholar]
- 4.Ma X, Chow JM, Cri G, et al. The interleukin 12 p40 gene promoter is primed by interferon-γ in monocytic cells. J Exp Med. 1996;183:147–57. doi: 10.1084/jem.183.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flesch IEA, Hess JH, Huang S, et al. Early interleukin 12 production by macrophages in response to mycobacterial infection depends on interferon γ and tumour necrosis factor α. J Exp Med. 1995;181:1615–21. doi: 10.1084/jem.181.5.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monteleone G, Biancone L, Marasco R, et al. Interleukin 12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology. 1997;112:1169–78. doi: 10.1016/s0016-5085(97)70128-8. [DOI] [PubMed] [Google Scholar]
- 7.Parronchi P, Romagnani P, Annunziato F, et al. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn's disease. Am J Pathol. 1997;150:823–32. [PMC free article] [PubMed] [Google Scholar]
- 8.Pallone F, Fais S, Boirivant M. The interferon system in inflammatory bowel disease. In: Fiocchi C, editor. Cytokines in inflammatory bowel disease. Austin, Texas: R G Landes Co.; 1995. pp. 57–67. [Google Scholar]
- 9.Fais S, Capobianchi MR, Pallone F, et al. Spontaneous release of interferon gamma by intestinal lamina propria lymphocytes in Crohn's disease. Kinetics of in vitro response to interferon gamma inducers. Gut. 1991;32:403–7. doi: 10.1136/gut.32.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fais S, Capobianchi MR, Silvestri M, et al. Interferon expression in Crohn's disease patients: increased interferon-gamma and -alpha mRNA in the intestinal lamina propria mononuclear cells. J Interferon Res. 1994;14:235–8. doi: 10.1089/jir.1994.14.235. [DOI] [PubMed] [Google Scholar]
- 11.Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-γ, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–70. [PubMed] [Google Scholar]
- 12.Neurath MF, Fuss I, Kelsall BL, et al. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–90. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monteleone G, Parrello T, Luzza F, et al. Response of human intestinal lamina propria T lymphocytes to interleukin 12: additive effects of interleukin 15 and 7. Gut. 1998;43:620–8. doi: 10.1136/gut.43.5.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhn R, Lohler J, Rennick D, et al. Interleukin-10 deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 15.Sellon RK, Tonkonogy S, Schultz M, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–31. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Portanova JP, Zhang Y, Anderson GD, et al. Selective neutralization of prostaglandin E2 blocks inflammation, hyperalgesia, and interleukin 6 production in vivo. J Exp Med. 1996;184:883–91. doi: 10.1084/jem.184.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams TJ, Morley J. Prostaglandins as potentiators of increased vascular permeability in inflammation. Nature. 1973;246:215–7. doi: 10.1038/246215a0. [DOI] [PubMed] [Google Scholar]
- 18.Phipps RP, Stein SH, Roper RL. A new view of prostaglandin E regulation of the immune response. Immunol Today. 1991;12:349–52. doi: 10.1016/0167-5699(91)90064-Z. [DOI] [PubMed] [Google Scholar]
- 19.van der Pouw Kraan TCTM, Boeije LCM, Smeenk RJT, et al. Prostaglandin-E2 is a potent inhibitor of human interleukin 12 production. J Exp Med. 1995;181:775–9. doi: 10.1084/jem.181.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strassmann G, Koota VP, Finkelman F, et al. Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J Exp Med. 1994;180:2365–70. doi: 10.1084/jem.180.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheuer WV, Hobbs MV, Weigle WO. Interference with tolerance induction in vivo by inhibitors of prostaglandin synthesis. Cell Immunol. 1987;104:409–16. doi: 10.1016/0008-8749(87)90042-6. [DOI] [PubMed] [Google Scholar]
- 22.Rugtveit J, Nilsen EM, Bakka A, et al. Cytokine profiles differ in newly recruited and resident subsets of mucosal macrophages from inflammatory bowel disease. Gastroenterology. 1997;112:1493–505. doi: 10.1016/s0016-5085(97)70030-1. [DOI] [PubMed] [Google Scholar]
- 23.Nathens AB, Rotstein OD, Dackiw APB, et al. Intestinal epithelial cells down-regulate macrophage tumor necrosis factor-alpha secretion: a mechanism for immune homeostasis in the gut-associated lymphoid tissue. Surgery. 1995;118:343–51. doi: 10.1016/s0039-6060(05)80343-5. [DOI] [PubMed] [Google Scholar]
- 24.Young HA, Hardy KJ. Role of interferon-gamma in immune cell regulation. J Leuk Biol. 1995;58:373–81. [PubMed] [Google Scholar]
- 25.Hayes MP, Wang J, Norcross MA. Regulation of interleukin-12 expression in human monocytes: selective priming by interferon-γ of lipopolysaccharide-inducible p35 and p40 genes. Blood. 1995;86:646–50. [PubMed] [Google Scholar]
- 26.Docke W-D, Randow F, Syrbe U, et al. Monocyte deactivation in septic patients: restoration by IFN-γ treatment. Nature Med. 1997;3:678–81. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 27.Beutler B, Tkacenko V, Milsark I, et al. Effect of γ interferon on cachetin expression by mononuclear phagocytes. Reversal of the lpsd (endotoxin resistance) phenotype. J Exp Med. 1986;164:1791–6. doi: 10.1084/jem.164.5.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Andrea A, Ma X, Aste-Amezaga M, et al. Stimulatory and inhibitory effects of interleukin (IL)-4 and IL-13 on the production of cytokines by human peripheral blood mononuclear cells: priming for IL-12 and tumor necrosis factor α production. J Exp Med. 1995;181:537–46. doi: 10.1084/jem.181.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li C, Goodrich JM, Yang X. Interferon-gamma (IFN-γ) regulates production of IL-10 and IL-12 in human herpesvirus-6 (HHV-6)-infected monocyte/macrophage lineage. Clin Exp Immunol. 1997;109:421–5. doi: 10.1046/j.1365-2249.1997.4661362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takasuka N, Tokunaga T, Akagawa KS. Preexposure of macrophages to low doses of lipopolysaccharide inhibits the expression of tumor necrosis factor-α mRNA but not of IL-1β mRNA. J Immunol. 1991;146:3824–30. [PubMed] [Google Scholar]
- 31.Madara JL, Stafford J. Interferon-γ directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest. 1989;83:724–7. doi: 10.1172/JCI113938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams RB, Planchon SM, Roche JK. IFN-γ modulation of epithelial barrier function. Time course, reversibility, and site of cytokine binding. J Immunol. 1993;150:2356–63. [PubMed] [Google Scholar]
- 33.Strober W, Kelsall B, Fuss I, et al. Reciprocal IFN-γ and TGF-β responses regulate the occurrence of mucosal inflammation. Immunol Today. 1997;18:61–64. doi: 10.1016/s0167-5699(97)01000-1. [DOI] [PubMed] [Google Scholar]
- 34.Duchmann R, Schmitt E, Knolle P, et al. Tolerance towards resident intestinal flora in mice is abrogated in experimental colitis and restored by treatment with interleukin-10 or antibodies to interleukin-12. Eur J Immunol. 1996;26:934–8. doi: 10.1002/eji.1830260432. [DOI] [PubMed] [Google Scholar]
- 35.Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol. 1991;146:108–13. [PubMed] [Google Scholar]
- 36.Snijdewint FGM, Kalinski P, Wierenga EA, et al. Prostaglandin E2 differentially modulates cytokine secretion profiles of human T helper lymphocytes. J Immunol. 1993;150:5321–9. [PubMed] [Google Scholar]
- 37.Stewart TH, Hetenyi C, Rowsell H, et al. Ulcerative enterocolitis in dogs induced by drugs. J Pathol. 1980;131:363–78. doi: 10.1002/path.1711310408. [DOI] [PubMed] [Google Scholar]
- 38.Bjarnason I, Smethurst P, Levi AJ, et al. Indomethacin induced chronic small intestinal ulceration in the rat. Gastroenterology. 1988;94:1937. [Google Scholar]
- 39.Rieser C, Papesh C, Herold M, et al. Differential deactivation of human dendritic cells by endotoxin desensitisation: role of tumor necrosis factor-α and prostaglandin E2. Blood. 1998;91:3112–7. [PubMed] [Google Scholar]