

1 INTRODUCTION
Polyclonal immunoglobulin, used for replacement therapy in immune deficiencies, must contain the full range of protective antibodies in order to provide prophylaxis against infections. Unlike Factor VIII, there is no possibility that this therapy could be provided solely by recombinant technology. Immunoglobulin for immunodeficient patients will continue to be produced from large pools of human plasma which maximizes protection but increases the chance that a unit of plasma contaminated with an infectious agent will be included.
For clinicians to ensure that benefit to an individual immunodeficient patient is maximal, this life-long therapy must be as safe as possible. Recently, immunoglobulin products have been withdrawn and plasma donors restricted in attempts to prevent transmission of blood-borne agents. Such measures have resulted in alarming shortages of therapeutic immunoglobulin worldwide. 1 This balance must be redressed by a critical look at the evidence regarding transmission of the wide range of blood-borne diseases potentially contaminating immunoglobulin for replacement therapy. The safety of replacement and high-dose therapies in relation to infusion-related reactions is not included here.
There are several methods by which immunoglobulin preparations are produced from human plasma and each product is generically different. Although efficacy is equivalent between these products, there are important differences which impinge on their long-term safety. Worldwide, there are currently over 25 preparations of immunoglobulin for use intravenously and more than six preparations used subcutaneously or intramuscularly. Almost all are produced by initial processing of pooled human plasma (from 1000 to 10 000 donors) by cold ethanol precipitation (Cohn–Oncley procedure),2 resulting in five plasma fractions. Cohn fraction II provides a preparation appropriate for intramuscular and subcutaneous use and is the starting material for purification of immunoglobulin for intravenous use by a variety of methods. Blood-borne agents have the potential to contaminate immunoglobulin, and therefore additional antiviral steps are used, before or after the Cohn–Oncley procedure, to reduce these risks. As the evidence for viral transmission by immunoglobulin is fragmentary, recommendations for a more systemic method of data collection are made so that real risk–benefit assessments for immunodeficient patients can be ascertained.
2 TYPES OF TRANSMISSABLE ORGANISMS
The types of transmissable organisms are discussed in order of their relevance to safety of immunoglobulin (see Table 1). Although blood can be contaminated by bacteria and protozoa, blood-borne viruses are the major concern because bacteria and protozoa are unlikely to survive the cold ethanol precipitation procedure used to produce immunoglobulin.
Table 1.
Ease of transmission via immunoglobulin therapy

Hepatitis B virus was a major problem in the 1970s but the development of appropriate HBV screening assays has eliminated transmission of HBV3 in immunoglobulin, provided that standards of production and quality assurance of assays are maintained. In the last 15 years there have been new concerns: human immunodeficiency viruses (HIV) 1 and 2; hepatitis C, with transmission via several immunoglobulin preparations;3 Creutzfeldt–Jakob disease (CJD); and, most recently, variant CJD.
2.1 Human immunodeficiency viruses 1 and 2 (HIV)
Retroviruses are inactivated by the cold alcohol precipitation, which is used universally in the manufacture of immunoglobulin. This fortuitous finding of reduced infectivity, along with the partitioning which takes place with each fractionation step,4,5 probably explains why transmission of HIV1 or 2 by immunoglobulin has not been confirmed, despite surveillance.6 The ongoing screening of donor units for HIV antibodies, combined with donor questionnaires regarding risk categories, remains essential.
2.2 Hepatitis C virus (HCV)
HCV is a lipid-coated virus with a viral core of approximately 33 nm. It is present in high concentrations early in the disease, prior to the detection of HCV antibodies (the ‘window period’).7 Contamination of donor blood is therefore not always detected by the antibody-based screening methods used at present and HCV may be present in the plasma pools from which immunoglobulin is subsequently purified.
Transmission of HCV by immunoglobulin has been reported 10 times since 1984,3 involving almost 4000 patients worldwide although this may be an underestimate. The new antiviral measures of pasteurization, nanofiltration or solvent detergent treatment, added to the manufacturing procedures recently, reduce this risk because the lipid nature of the virus coat makes it susceptible to detergent treatment and the size of the virus enables removal by nanofiltration. Parallels with factor 8 suggest that these steps have reduced HCV transmission in haemophiliac patients but only continuing surveillance will show whether these additional methods, proven on surrogate lipid-coated viruses, are equally effective for immunoglobulin. Statutory documentation of product and lot numbers of immunoglobulin would enable tracing of patients retrospectively (as for HCV in blood transfusion).
2.3 Creutzfeldt–Jakob diseases (CJD)
Creutzfeldt–Jakob disease (CJD) is one of the transmissible spongiform encephalopathies (TSEs), a group of degenerative brain diseases that affect animals and humans. TSE in animals includes scrapie in sheep, bovine spongiform encephalopathy (BSE) in cows and kuru and CJD in humans.
Kuru was associated with cannibalism and was transmitted orally; how the presumed infective particles moved from the site of entry to the brain remains speculative. It was transferred to chimpanzees by intracerebral injection of affected human brain, but there was no evidence that kuru was transmitted by blood.
CJD is a rare condition with an incidence of 0.5–1 people per million of population per year (Table 2). There are three well-recognized forms of CJD: sporadic (spCJD), iatrogenic and familial CJD. Patients with spCJD are usually between the ages of 50 and 70 years, have a rapidly progressive mental deterioration with myoclonus and a typical EEG pattern. There is a long preclinical period but, once diagnosed, patients die within a few months. Iatrogenic CJD (< 1% of human cases) has followed transplantation of cornea or dura mater or injections of pituitary growth hormone derived from cadavers.
Table 2.
Deaths of definite and probable cases of Creutzfeldt–Jakob disease (CJD) in the UK, 1985–1999
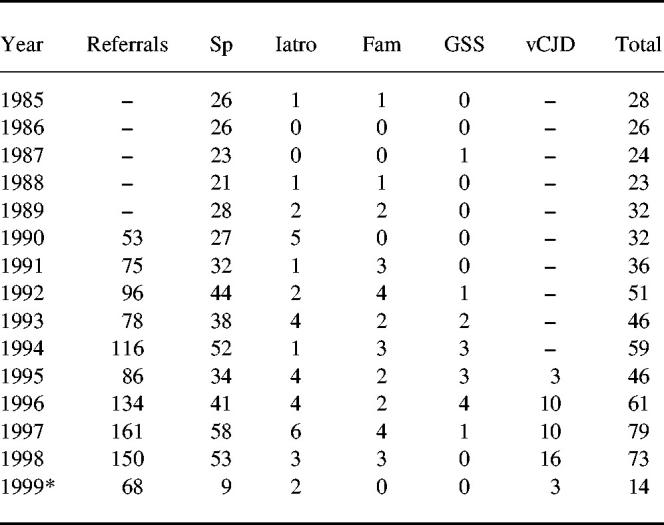
Data from CJD surveillance unit, Edinburgh, UK.
SP, sporadic CJD; Iatro = iatrogenic CJD; Fam = familial CJD
GSS, Gertmann–Straussler–Scheinher syndrome.vCJD, variant CJD.
to 31.5.99.
The prion protein is a normal, protease-sensitive, component of cells present in high concentration in the normal central nervous system and in many other organs including lymphoid tissue. Little is know of its physiological role. TSEs are characterized by deposition within the brain of an abnormal protease-resistant form of the prion protein. Inoculation of material containing abnormal prion protein into the brains of experimental animals may result in the appearance of the typical plaques of spongiform degeneration.
Transmission, both accidental and experimental, has been shown for some prions. Most is known about the agent of scrapie which has been shown to be transmissible in rodents from infected spleens (via follicular dendritic cells and/or by activated B cells), as well as from brain, demonstrating extraneuronal infection and a possible route of transmission. For CJD, retrospective studies of recipients of blood transfusions from donors who later died of spCJD have found no cases.8 Case control studies have shown no evidence of excess blood transfusions in those with spCJD9 and there has been no transmission of spCJD following infusion of blood to three monkeys over 16 years of follow-up.10 Autopsies on the brains of haemophiliacs have found none of the pathological signs of spCJD so far: these studies are continuing in view of the long incubation period of spCJD.11 Although there are, at present, no data on the risk of transmission of CJD by immunoglobulin, these postmortem studies should be extended to all recipients of immunoglobulin (antibody deficient and immunocompetent) in order to provide this important evidence. Early referral to surveillance units of immunoglobulin recipients who develop unexplained neurological or psychiatric symptoms, especially those on long-term therapy for immune deficiencies, is essential.
Concerns about these diseases have resulted, in some countries, in the recall of those batches of immunoglobulin containing plasma from a donor subsequently developing, or at risk of developing, CJD. In 1996/7, 15% of all IVIg produced in the USA was withdrawn as a result of such recall. The risk should be compared with the high risk to immunodeficient patients without replacement immunoglobulin, for whom there is no alternative therapy. It is important that regulators consult immunodeficient patients' representatives and their medical advisers in order to assess the risks to this particular group of patients who require life-long immunoglobulin treatment.
2.4 Variant CJD
The possibility of transmission of BSE from cattle to humans by consumption of beef was raised in 1986. The BSE epidemic in cows, which rose to a peak in 1992 when 36 682 confirmed cases were reported, is gradually coming under control with only approximately 3100 confirmed cases in the UK in 1998; there have been few reported cases in other countries. A causal link with variant CJD was raised by the successful cerebral transmission of BSE into macaque monkeys, resulting in neuropathy similar to that in patients with vCJD.12 Variant CJD is distinct from spCJD, with a different clinical presentation (depression and abnormal sensory symptoms), the absence of the typical EEG and a younger age at diagnosis. Conclusive evidence that the agents for BSE and vCJD were identical came from two studies in 1997.13,14 Disease-specific prions (PrP vCJD) have since been detected in lymphocytes in tonsil and appendix, raising concern about transmissibility in blood or blood products.15,16
In view of the theoretical risk that circulating lymphoid cells might transmit vCJD, the UK Government decided not to use UK-derived plasma for production of plasma concentrates including immunoglobulin, thus reducing the availability of plasma for manufacture of immunoglobulin. However, numbers of cases of vCJD have remained low over 3 years (Table 2) and there is, as yet, no evidence of human-to-human transmission.17 Six of the 40 vCJD cases were known to have given blood prior to the development of symptoms; the plasma was used in the production of albumin rather than immunoglobulin and tracing has not been informative so far. Postmortem brain studies (as for spCJD) of immunoglobulin recipients would provide data to monitor any transmission by immunoglobulin therapy.
2.5 Viruses not relevant to immunoglobulin
Hepatitis A virus (HAV) is principally transmitted orally. It is a small nonenveloped virus with remarkable homogeneity; antibodies directed against one strain are protective against more than 90% of other strains. However, HAV has been transmitted in solvent detergent-treated Factor VIII, and immunoglobulin contains relatively high levels of neutralizing antibodies and has not been shown to transmit hepatitis A; some regulators still insist on a minimum level of HAV antibodies for intramuscular immunoglobulins.
Parvovirus B19, which causes a mild self-limiting illness in children but is also associated with transient red cell aplasia, is a small virus, resistant to heat and detergent. Seroconversion has been shown after dry and steam-treated Factor VIII therapy, but high levels of specific antibodies in immunoglobulin (associated with seropositivity in at least 50% of adults) are presumed to neutralize any persistent virus because normal immunoglobulin preparations have been used to ameliorate and to resolve B19 parvovirus infections.
Hepatitis G virus (HGV) is a newly discovered virus detected in patients with acute or chronic hepatitis, especially those with HCV. However, in a study of HCV antibody-negative liver transplant recipients there was no difference in the subsequent incidence of hepatitis between those with and those without HGV infection.18 Reassurance that HGV is not clinically relevant was shown by a low prevalence of HGV RNA in patients with end-stage liver disease.19,20 A novel DNA virus, transfusion transmitted virus (TTV), which has a high incidence in blood donors and patients with chronic liver disease, also appears to have no clear disease association.21,22
2.6 Unknown agents
Continued monitoring of the field of emerging pathogens is required to ensure safety against new viruses or other agents. Storage of serum specimens, pre-treatment and 1 month later when commencing therapy or changing immunoglobulin products, as well as serially (e.g. every 6 months) for those on chronic therapy, would enable detection of new agents when assays become available.
3 PLASMA COLLECTION
3.1 Choice of donors
The plasma donor population is important; the choice depends on the prevalences of the relevant blood-borne diseases. Collection from donors in countries where there is a high prevalence of blood-borne diseases is now avoided and data from collection centres in the early 1990s showed low prevalence rates for HIV, HBV and HCV.23
In the early 1970s transfusion-related hepatitis C was less frequently associated with volunteer blood compared with blood from paid donors, leading to the belief that blood was safer from unpaid donors. The issue of relative safety of plasma from volunteer (but often compensated) donors vs. paid donors has been revisited; there is a higher incidence of blood-borne infection markers in whole blood donors (usually unpaid), due to a higher level of HCV antibodies.24
Frequency of donation has also been controversial. Frequent donations may have contributed to HCV transmission by one specific lot of immunoglobulin in the most recent outbreak.25 Donation frequency can be limited in order to prevent multiple collections of plasma during a ‘window’ period. Alternatively, donations can be held in ‘quarantine’ for a few months until the donor has been rechecked, enabling contaminated donations to be identified and destroyed. Studies on the actual prevention of transmission by quarantine is not yet available although extrapolation from serial testing and comparison of first-time donors with all donations24 provide indirect evidence for the success of such a strategy. The use of previously tested donors reduces the prevalence of ‘positive markers’ and hence the need to recall batches of immunoglobulin retrospectively if a donation is subsequently positive.
3.2 Accreditation of blood collection centres and manufacturing plants
In the late 1980s, concerns about the standards of blood and plasma collection led to rigorous improvements, with obligatory good manufacturing practice protocols and quality assurance schemes. Facilities for collection and fractionation of plasma are inspected and may be suspended if the centre does not adhere to the standards.
Failure of manufacturers to comply with the strict application of modern accrediting standards and procedures, and the insistence of regulators to repeat inspections each time a change is made in the processing of immunoglobulin, have resulted in temporary closure of manufacturing plants, contributing to the worldwide shortages. For example, these two factors probably accounted for 60% of the immunoglobulin shortages in the US in 1998.
3.3 Individual screening of donors and the donated units of plasma
Questionnaires, designed to screen out donors whose medical, behavioural or travel histories indicate an infection risk, are used worldwide; questions relating to CJD and vCJD are now included in the questionnaire. The shortcomings of this process were highlighted by the finding that 1.7% of all donors were prepared to report risk behaviour only when asked anonymously.24 This emphasizes the need for in vitro screening of all donated units of blood and plasma.
Antigen testing is probably the screening method of choice for blood donations because there have been no cases of HBV transmission by immunoglobulin since routine HBV surface antigen screening was introduced. There are currently no specific antigen tests for HCV, spCJD or vCJD, although assays are being developed; HIV antigen screening is now used by some blood collection agencies.
The shortcomings of antibody testing have been highlighted. The sensitivity and specificity of the assays are also important, particularly as enzyme-linked immunosorbent assays (ELISA) are the standard method of detection. Although the currently used ELISAs for HIV1 and HIV2 have sensitivities and specificities of > 99%, those for HCV caused problems when plasma screening for HCV antibodies was first adopted. There were concerns that removal of these antibodies from plasma pools would compromise the safety of immunoglobulin26 but immunoglobulin made from such plasma did not transmit HCV to chimpanzees and therefore regular screening was introduced, with the first lots of IVIg available in mid-1993. Initially, an antibody screening assay using a single recombinant antigen was used which gave a high proportion of false-negative results and a product which apparently did not transmit HCV.27 The introduction of ‘an improved test’ using several antigens, with greater efficiency of removal of HCV antibodies, was followed by an outbreak of hepatitis C in over 200 immunoglobulin recipients in late 1993–1994. Subsequent studies showed that several lots of immunoglobulin had high levels of HCV-RNA by polymerase chain reaction (PCR).28 It was surprising that more than half the commercial intramuscular immunoglobulin preparations were also HCV-RNA positive, despite the absence of HCV transmission by this type of immunoglobulin. PCR methods detect viral nucleic acid during the viraemic stage and could be used to overcome the ‘window’ periods in HCV and HIV. Some manufacturers are PCR-testing smaller plasma pools (to prevent loss of plasma if contamination is demonstrated) but HCV-PCR testing of the individual donor units might be needed.29 Evidence is lacking at present and serial testing of donors and quarantining of donations may prove to be satisfactory.
4 MANUFACTURING PROCESSES
4.1 Plasma fractionation
Plasma fractionation relies on the varying solubility of plasma proteins according to pH, temperature and alcohol concentration. At each stage the insoluble proteins are separated by centrifugation and viruses can contaminate each of the insoluble precipitates. Reductions in the viral load have been demonstrated by serial partitioning, for both HCV30 and HIV.31 However, the role of partitioning of viruses cannot be taken in isolation; when antibodies to HCV were removed following the introduction of screening, the amount of recoverable HCV-RNA in the various Cohn fractions changed dramatically as a result of the virus no longer being complexed with antibody.28
4.2 Downstream processing
Fraction II provides immunoglobulin for replacement therapy by the intramuscular or subcutaneous routes and only requires further purification if it is to be used intravenously. A number of different methods are used, including ion exchange chromatography, acid pH 4 with or without additional enzymes or polyethylene glycol precipitation. It was discovered, again retrospectively, that the process of acidification, with or without enzymes, has the added benefit of inactivating HCV and its surrogate virus, bovine vesicular diarrhoea virus (BVDV).32
4.3 Viral inactivation methods
As Cohn–Oncley fractionation is not sufficient to remove lipid-coated viruses, additional antiviral inactivation steps are required. Heating (steam) is used for immunoglobulin but the molecules tend to aggregate at 60°C unless stabilized. Solvent detergent steps are increasingly popular.33 Nanofiltration, with pore sizes of 15 or 35 nm, appears to offer a logical (although technically difficult) method to remove larger viruses but is unlikely to remove nonfibrillar prions. The regulators have issued guidelines for validation of these procedures, which include proof of viral inactivation and relevance of surrogate viruses.
5 MONITORING OF PRODUCT USAGE
Regulators and manufacturers have paid insufficient attention to collecting safety data by monitoring recipients of immunoglobulin. Given the statutory requirements for careful documentation of the use of blood, the failure to insist on monitoring of the use of soluble blood products is extraordinary. This lack of information makes tracing of transmission extremely difficult; for example, in the recent 1994 outbreak of hepatitis C, usage of specific batches of immunoglobulin was identified in only 70% of recipients in Europe and 50% in the USA. In contrast, where there were full data on batch usage, prompt detection of transmitted infection34 and early treatment with interferon alpha were possible; early treatment was shown to be more effective than treatment in the chronic phase.35,36
Post-licensing monitoring by manufacturers is obligatory; manufacturers are required to keep aliquots of each batch of immunoglobulin produced so that an individual virus can be traced retrospectively by genotyping. This allows good epidemiological investigation, both within a patient population and, more importantly, to identify a contaminated donor or an unsafe manufacturing process.
6 CONCLUSION
The concerns of regulators to ensure that transmission of all blood-borne diseases be kept to an absolute minimum is important but the principle of precaution on which decisions have been based worldwide has caused severe shortages of immunoglobulin.1 In order to balance the safety of immunoglobulin replacement therapy against the morbidity of immunodeficient patients with inadequate replacement, there needs to be greater consistency of worldwide regulatory agencies, more involvement of users (physicians and patients) and collection of more data relating to precise risk. Issues include the size of plasma pools, methods of testing for blood-borne agents, continuing research on possible transmission of pathogens and refinement of viral inactivation methods.
Another problem relating to availability is the ever-widening list of indications for immunoglobulin, particularly in high doses in autoimmune conditions and Kawasaki disease. The expected increase in demand is 20% per year although supplies are expected to increase by only 10% pa. Purchasers of healthcare (governmental and private) should be encouraged to audit the use of this scarce resource and every effort for alternative products, including the development of targeted therapies for nonimmunodeficient patients, should be sought. Removal of regulatory restraints to the movement of plasma and its derivatives around the world is essential if these life-saving therapies are to continue to be available to all the patients who need them.
RECOMMENDATIONS FOR THE SAFE USE OF THERAPEUTIC IMMUNOGLOBULIN
Statutory documentation of the name of the product and lot numbers used in individual patients for traceability.
Required monitoring by physicians, i.e. assays for detection of antibodies to HCV (with HCV-PCR in antibody deficiency patients) and liver function tests in all recipients of immunoglobulin. This should be done pretherapy and serially, including 6 months after the last immunoglobulin dose.
Storage of serum specimens, pre-treatment and 1 month later when commencing therapy or changing products, and serially (e.g. every 6 months) for chronic therapy, to enable retrospective detection of any new agents.
Early referral to surveillance units of recipients of immunoglobulin, especially those on long-term, repeated therapy, if they develop unexplained neurological or psychiatric symptoms.
Consideration of establishing programmes for registration of recipients receiving repeated doses of immunoglobulin for eventual postmortem brain examination.
Establishment of national or regional registers of those individuals infected by immunoglobulin.
Greater consistency of worldwide regulatory agencies and the wider involvement of users including immunodeficient patients and their physicians.
Encouragement of partnership between manufacturers, patients' representatives and prescribers to improve consistency of information and data/evidence relating to risk: benefit analysis for gaining informed consent.
In view of the short supply of immunoglobulin, encouragement for development of alternative products, including targeted therapies for nonimmunodeficient patients.
Consideration of a common policy for deferring previous blood/blood components/blood products recipients from future donation of blood for use in plasma pools from which immunoglobulin is processed.
References
- 1.Milgrom H. Shortages of intravenous immunoglobulin. Ann Allergy, Asthma Immunol. 1998;81:97–9. doi: 10.1016/S1081-1206(10)62792-5. [DOI] [PubMed] [Google Scholar]
- 2.Cohn EJ, Strong LE, Hughes WL. Preparation and properties of serum and plasma proteins IV. A system for the separation into fractions of the protein and lipoprotein components of biological tissues and fluids. Ann Am Chem Soc Year. 1946;68:459–7. doi: 10.1021/ja01207a034. [DOI] [PubMed] [Google Scholar]
- 3.Yap PL. Viral Safety of IVIg. In: Lee M, Stand V, editors. Intravenous Immunoglobulin in Clinical Practice. 1997. pp. 67–10. Marcel Dekker. [Google Scholar]
- 4.Mitra G, Wong MF, Mozen MM, McDougal FS, Levy JA. Elimination of infectious retroviruses during preparation of immunoglobulin. Transfusion. 1986;26:394–7. doi: 10.1046/j.1537-2995.1986.26486262753.x. [DOI] [PubMed] [Google Scholar]
- 5.Wells MA, Wittek AE, Epstein JS. Inactivation and partition of human T cell lymphotrophic virus, type III, during ethanol fractionation of plasma. Transfusion. 1986;26:210–3. doi: 10.1046/j.1537-2995.1986.26286152919.x. [DOI] [PubMed] [Google Scholar]
- 6.Center for Disease Control. Safety of therapeutic immunoglobulin presss with respect to transmission of human T lymphocyte virus type I/lymphadenopathy associated virus infection. MMWR. 1986;35:231–2. [Google Scholar]
- 7.Vrielink H, Vander Poel CL, Reesink HW, Zaaijer HC, Lelie PN. Transmission of HCV by anti-HCV negative blood transfusion. Vox Sang. 1995;68:55–6. doi: 10.1111/j.1423-0410.1995.tb02546.x. [DOI] [PubMed] [Google Scholar]
- 8.Heye N, Hensen S, Muller N. spCJD in blood transfusion. Lancet. 1994;343:298–9. doi: 10.1016/s0140-6736(94)91148-7. [DOI] [PubMed] [Google Scholar]
- 9.Wientjens DPWM, Davimipour Z, Hofman A, Kando K, Matthews WB, Hill RG, Van Duij CM. Risk factors for CJD. A re-analysis of case control studies. Neurology. 1996;46(5):1287–9. doi: 10.1212/wnl.46.5.1287. [DOI] [PubMed] [Google Scholar]
- 10.Brown P, Gibbs CJ, Rodgers-Johnson P. Human spongioform encephalopathy: the NIH series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;44:513–5. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 11.Lee CA, Ironside JW, Bell JE, Giangrande P, Ludlam C, Esiri MM, McLaughlin JE. Retrospective neuropathologial review of prion disease in UK haemophiliac patients. Thromb Haemost. 1998;80:909–1. [PubMed] [Google Scholar]
- 12.Lasmezas CI, Deslys J, Demalmay R. BSE transmission to macaques (letter) Nature. 1996;381:743–4. doi: 10.1038/381743a0. [DOI] [PubMed] [Google Scholar]
- 13.Bunce ME, Hill RG, Ironside JW. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–50. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 14.Hill AF, Destruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey LJ. Lantos. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–5. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 15.Hill AF, Zeidler M, Ironside J, Collinge J. Diagnosis of new variant Creutzfeldt–Jakob disease by tonsil biopsy. Lancet. 1997;349((9045)):99–10. doi: 10.1016/S0140-6736(97)24002-X. [DOI] [PubMed] [Google Scholar]
- 16.Hilton DA, Fathers E, Edwards P, Ironside J, Zajicek J. Prion immunoreactivity in appendix before the clinical onset of variant Creutzfeldt–Jakob disease. Lancet. 1998;352:703–4. doi: 10.1016/S0140-6736(98)24035-9. [DOI] [PubMed] [Google Scholar]
- 17.Will RG, Cousens SN, Farrington CP, Smith PG, Knight RSG, Ironside JW. Deaths from variant Creutzfeldt–Jakob disease. Lancet. 1999;353:979–8. doi: 10.1016/s0140-6736(99)01160-5. [DOI] [PubMed] [Google Scholar]
- 18.Hoofnagle JH, Lombardero M, Wei Y. Hepatitis G virus infection before and after liver transplantation. Liver Transpl Surg. 1997;3:578–8. doi: 10.1002/lt.500030604. [DOI] [PubMed] [Google Scholar]
- 19.Alter HJ, Nakatasugi Y, Mea J. The incidence of transfusion associated hepatitis G virus infection and its relation to liver disease. New Eng J Med. 1997;336:747–5. doi: 10.1056/NEJM199703133361102. [DOI] [PubMed] [Google Scholar]
- 20.Laskus T, Wang LF, Redknowski M. Hepatitis G virus infection in American patients with cryptogenic cirrhosis: no evidence for liver replication. J Infect Dis. 1997;176:1491–5. doi: 10.1086/514146. [DOI] [PubMed] [Google Scholar]
- 21.Simmonds P, Davidson S, Lycett C. Detection of a novel DNA virus (TTV) in blood donors and blood products. Lancet. 1998;352:191–5. doi: 10.1016/s0140-6736(98)03056-6. [DOI] [PubMed] [Google Scholar]
- 22.Naoumov NV, Petrova EP, Thomas MG, Williams R. Presence of a newly described human DNA virus (TTV) in patients with liver disease. Lancet. 1998;352:195–7. doi: 10.1016/S0140-6736(98)04069-0. [DOI] [PubMed] [Google Scholar]
- 23.Westphal RG. Donors and the US blood supply. Transfusion. 1997;37:237–4. doi: 10.1046/j.1537-2995.1997.37297203531.x. [DOI] [PubMed] [Google Scholar]
- 24.Glynn SA, Schreiber GB, Busch MP. Demographic characteristics, unreported risk behaviour and prevalence and incidence of viral infections: a comparison of apheresis and whole blood donors. Transfusion. 1998;38:350–8. doi: 10.1046/j.1537-2995.1998.38498257373.x. [DOI] [PubMed] [Google Scholar]
- 25.Gomperts ED. Gammagard and reported HCV episodes. Clin Therapeutics. 1996;18(Suppl. B):3–8. doi: 10.1016/s0149-2918(96)80192-5. [DOI] [PubMed] [Google Scholar]
- 26.Finlayson J, Tankersley DL. Anti-HCV screening and plasma fractionation: the case against. Lancet. 1990;335:1274–5. doi: 10.1016/0140-6736(90)91335-8. [DOI] [PubMed] [Google Scholar]
- 27.Biswas RM, Nedjar S, Wilson LT. The effect on the safety of intravenous imunoglobulin of testing plasma for antibody to hepatitis C. Transfusion. 1994;34:100–4. doi: 10.1046/j.1537-2995.1994.34294143934.x. [DOI] [PubMed] [Google Scholar]
- 28.Yu MW, Mason BL, Tankersley DL. Detection and quantitation of HCV RNA in immune globulins produced by Cohn-Oncley fractionation of human plasma. Transfusion. 1994;34:596–60. [Google Scholar]
- 29.Schottstedt V, Tuma W, Bunger G, Lefevre H. PCR for HBV, HCV and HIV-1: first results from a routine screening programme in a large blood transfusion service. Biologicals. 1998;26(2):101–4. doi: 10.1006/biol.1998.0144. [DOI] [PubMed] [Google Scholar]
- 30.Yei S, Yu MW, Tankersley DL. Partitioning of hepatitis C virus during Cohn-Oncley fractionation of plasma. Transfusion. 1992;32:824–8. doi: 10.1046/j.1537-2995.1992.32993110753.x. [DOI] [PubMed] [Google Scholar]
- 31.Hart H, Macomish F, Hart WG, Simmons P, Transfusion PLY. A comparison of PCR with an infectivity assay for HIV-1 titration during the virus inactivation of blood products. Transfusion. 1993;33:838–4. doi: 10.1046/j.1537-2995.1993.331094054622.x. [DOI] [PubMed] [Google Scholar]
- 32.Louie REJ, Galloway C, Dumas ML. Inactivation of hepatitis C virus in low pH intravenous immunoglobulin. Biologicals. 1994;22:13–9. doi: 10.1006/biol.1994.1003. [DOI] [PubMed] [Google Scholar]
- 33.Horowitz B. Preparation of virus sterilized immune globulin solutions by treatment with organic solvent-detergent mixtures. In: Krijnen HW, Strengers PFW, Aken WG, editors. Immunoglobulins. Amsterdam: Central Labortory of the Netherlands Red Cross Blood Transfusion Service; 1988. [Google Scholar]
- 34.Healey CJ, Sabharwal NK, Daub J. Outbreak of acute hepatitis C following the use of anti-hepaitis C virus-screened intravenous immunoglobulin therapy. Gastroenterology. 1996;110:1120–6. doi: 10.1053/gast.1996.v110.pm8613001. [DOI] [PubMed] [Google Scholar]
- 35.Christie JML, Healey CJ, Watson J. Clinical outcome of hypogamma-globulinaemic patients following outbreak of acute hepatitis C. two year follow up. Clin Exp Immunol. 1997;110:4–8. doi: 10.1046/j.1365-2249.1997.5081412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bjoro MD, Froland SS, Yun Z, Samdal HH, Haaland MD. Hepatitis C infection in patients with primary hypogammaglobulinaemia after treatment with contaminated immune globulin. N Eng J Med. 1994;331:1607–1. doi: 10.1056/NEJM199412153312402. [DOI] [PubMed] [Google Scholar]