

Abstract
The ability of antineutrophil cytoplasm autoantibodies (ANCA) from patients with systemic vasculitis to stimulate protein kinase C (PKC) and tyrosine kinases was examined in human neutrophils. Using the superoxide dismutase-inhibitable reduction of ferricytochrome C, the kinetics of ANCA-induced superoxide (O2−) production were characterized and subsequently manipulated by specific inhibitors of PKC and tyrosine kinases. With this approach, ANCA IgG, but not normal IgG or ANCA F(ab′)2 fragments caused a time and dose dependent release of O2− from TNF-α primed neutrophils. The kinetics of ANCA-induced O2− production showed an initial 10–15 min lag phase compared to the N-formyl-l-methionyl-l-leucyl-l-phenylalanine response, suggesting differences in the signalling pathways recruited by these two stimuli. Inhibitor studies revealed that ANCA-activation involved members of both the Ca2+-dependent and -independent PKC isoforms and also tyrosine kinases. ANCA IgG resulted in the translocation of the βII isoform of PKC at a time corresponding to the end of the lag phase of O2− production, suggesting that PKC activity may be instrumental in processes regulating the activity of the NADPH oxidase in response to ANCA. Tyrosine phosphorylation of numerous proteins also peaked 10–15 min after stimulation with ANCA but not normal IgG. These data suggest that PKC and tyrosine kinases regulate O2− production from neutrophils stimulated with autoantibodies from patients with systemic vasculitis.
Keywords: neutrophils, ANCA, tyrosine phosphorylation, PKC
INTRODUCTION
The primary small vessel vasculitides such as Wegeners granulomatosis (WG) and microscopic polyangiitis (MPA) are multisystem disorders characterized by inflammation of blood vessel walls and are an important cause of acute renal failure in adults [1]. WG and MPA are strongly associated with high levels of circulating antineutrophil cytoplasm autoantibodies (ANCA), autoreactive IgG antibodies directed towards myeloperoxidase (MPO) or proteinase 3 (PR3). A pathogenic role has been suggested for ANCA, with supporting evidence that, in vitro, ANCA are capable of directing neutrophil cytotoxicity towards cultured endothelial cells [2,3] and the ability of ANCA to activate primed neutrophils to degranulate and undergo a respiratory burst [4,5].
Current knowledge of the components of ANCA induced signal transduction leading to O2− production is limited. Studies by Lai and Lockwood [6,7] have suggested that ANCA activation of neutrophils leads to the translocation (an indication of activation) of PKC. A recent study [8] suggested that F(ab′)2 or cross-linked Fab fragments of ANCA are capable of initiating O2− production from cytokine primed neutrophils, whilst others suggest a role for Fcγ receptor engagement [9,10]. Controversy thus exists as to the mechanism of activation of neutrophils by ANCA.
Protein kinase C (PKC) is suggested to play a pivotal role in many signal transduction systems [11]. Eleven isoenzymes of PKC have been identified in mammalian tissues to date and can be divided into three groups according to their calcium and phospholipid requirements and their ability to be activated by the phorbol ester, 4β-phorbol 12-myristate 13-acetate (PMA) [12,13]. The conventional (‘c’) PKCs (α, βI, βII and γ) are calcium-dependent and activated by 1,2-diacylglycerol (DAG) or PMA. The novel (‘n’) PKCs (δ, ε, η and θ) are calcium-independent but responsive to DAG or PMA. Atypical (‘a’) PKCs [ζ and ι(λ)] are unresponsive to calcium, DAG or PMA, but, as with all isoforms of PKC, require phosphatidylserine for maximal activation. Differences in tissue distribution, intracellular location, cofactor requirement and enzymatic activity suggest a different role for each isoform of PKC in cellular function.
Neutrophils have been shown to express five PKC isoforms; α, βI and βII from the c-PKCs, δ from the n-PKCs and ζ from the a-PKCs [14–16]. It has been suggested that PKC may regulate/activate O2− production from the neutrophil NADPH oxidase system in response to external stimuli such as PMA or N-formyl-l-methionyl-l-leucyl-l-phenylalanine (fMLP) [17]. NADPH oxidase activation by fMLP has also been shown to be dependent on the activity of tyrosine kinases, possibly Lyn [18].
The purpose of this study was to examine signal transduction in neutrophils following ANCA IgG stimulation. In initial studies, the ability of ANCA to induce a respiratory burst from primed and unprimed neutrophils was examined and compared with that of known activators, namely fMLP and PMA. The effects of ANCA IgG and ANCA F(ab′)2 fragments used alone or cross-linked were compared to establish the surface receptor-binding requirements for activation. Subsequently, the role of PKC isoforms and tyrosine kinases in the ANCA-induced respiratory burst were examined using specific inhibitors, with confirmation of the activities of PKC and tyrosine kinases over time detected by Western blot analysis. Determination of the processes involved in ANCA activation of neutrophils should help us to gain a better insight into the mechanisms of autoimmunity.
MATERIALS AND METHODS
Isolation of neutrophils
Neutrophils were isolated using discontinuous plasma percoll gradients as described previously [19]. Neutrophils were resuspended at 2 × 106/ml in Dulbecco's phosphate buffered saline (PBS) (Sigma Chemical Co., Poole, Dorset, UK) prepared in pyrogen free water. Unless otherwise indicated, prior to all experiments the neutrophils were treated with 5 μg/ml cytochalasin B (Sigma) for 5 min at 37°C, followed by a priming dose of 2 ng/ml TNF-α (NISBC, London, UK) for 15 min. Neutrophils were 99% viable by trypan blue exclusion and > 98% pure by haematoxylin and eosin staining.
IgG isolation
IgG was isolated either from patient sera taken during active disease or from normal human sera by affinity chromatography using a HiTrap protein G affinity column (Pharmacia, Upsala, Sweden) using pyrogen free materials as described previously [20]. The specificity of anti-PR3 or anti-MPO ANCA was assessed by antigen specific ELISA and indirect immunofluorescence on ethanol-fixed neutrophils. All IgG samples were free of contaminating endotoxin as assessed by the limulus ameobocyte assay. Furthermore, neutrophil stimulation assays were carried out in the absence of serum to avoid binding and activation by endotoxin.
F(ab′)2 isolation
F(ab′)2 fragments were prepared by digestion with pepsin gel (Pierce, Chester, UK) in 20 mm sodium acetate buffer pH 4.0 for 16 h at 37°C. The reaction was terminated by neutralization with 10 mm Tris-HCl pH 7.5 and removal of the pepsin gel by centrifugation at 1000 g for 5 min. The preparations were then filtered (0.2 μ) to remove any traces of the pepsin gel. The contaminating Fc fragments and undigested whole IgG were removed using affinity chromatography on protein G and the remaining F(ab′)2 fragments dialysed against PBS. Purity of the F(ab′)2 fragments was assessed by SDS-PAGE and was always greater than 95% pure. Importantly, F(ab′)2 fragments prepared in this manner were still able to bind neutrophil antigens as assessed by both indirect immunofluorescence on ethanol fixed neutrophils and by antigen specific ELISA (data not shown). Three MPO-ANCA and two PR3-ANCA F(ab′)2 preparations were compared.
Superoxide generation
Superoxide production was determined discontinuously at 37°C using a kinetic microplate assay [21] with minor modifications. In short, 96-well plates were incubated with 50 μl neutrophil suspension (1 × 105 cells/well), 75 μm ferricytochrome c (Sigma) prepared in PBS containing 0.9 mm CaCl2, 0.5 mm MgCl2, 7.5 mm Glucose (PBSG), either 300 U/ml superoxide dismutase (SOD) (Sigma) or an equal volume of PBSG and stimulus. IgG preparations were added at a concentration of 100 μg/ml for MPO-ANCA and 200 μg/ml PR3-ANCA. fMLP and PMA, 1 μm and 100 ng/ml, respectively, served as positive controls. The plates were scanned at 10 min intervals over 100 min using a Multiscan® Bichromatic plate reader (Lifescienses, Hampshire, UK), with the plates maintained at 37°C between readings. O2− production was calculated using a molar extinction coefficient for ferricytochrome c of 21.1 × 103 m−1 cm−1 and a light path of 0.6 cm for a final well volume of 250 μl. Each test was performed in triplicate, on at least three different neutrophil donors.
Cross-linking studies
Neutrophils were resuspended to 2 × 106/ml in prewarmed PBSG. Following incubation with 2 ng/ml TNF-α for 15 min at 37°C, neutrophils were incubated in the presence or absence of ANCA F(ab′)2 fragments (150 μg/ml) for a further 15 min at 37°C with gentle agitation. The F(ab′)2 fragments were added at 150 μg/ml as this concentration was shown to give equivalent binding to 100 μg/ml of the whole IgG by antigen specific ELISA using doubling dilution. Unbound ANCA F(ab′)2 fragments were removed by washing in cold PBSG. The neutrophils were then resuspended to 2 × 106/ml in prewarmed PBSG and treated with 5 μg/ml cytochalasin B for 5 min at 37°C. O2− production was then measured in response to 100 μg/ml of an F(ab′)2 preparation of sheep antihuman IgG F(ab′)2 antibody (Binding Site, UK).
Inhibition of PKC
For the assessment of the involvement of PKC in neutrophil O2− production, inhibitors were added over a range of one log below to two logs above the published IC50 values for inhibition of PKC in an in-vitro assay system: Chelerytherine Chloride (IC50[PKC] = 0.66 μm [22]), Bisindolylmaleimide I (IC50 [cPKC − α, βI, βII and γ] ≅ 0.02 μm [23] [nPKC − δ] = 0.21 μm; [nPKC − ε] = 0.13 μm; [aPKC − ζ] = 5.8 μm [24]), Gö6976 (IC50 [cPKC − α, βI, βII and γ] = 2–10 nm [24]). In all cases cytochalasin B and TNF-α primed neutrophils were preincubated with inhibitor for 5 min at 37°C, before the addition of stimulus.
Inhibition of tyrosine kinases
Cytochalasin B and TNF-α primed neutrophils were preincubated for 30 min in the presence or absence of either 30 μm or 150 μm genistein, prior to addition of stimulus (IgG, fMLP or PMA).
Translocation of PKC: cell stimulation and fractionation
Human neutrophils were resuspended to 5 × 106/ml in prewarmed PBSG, treated with 5 μg/ml cytochalasin B for 5 min and primed with 2 ng/ml TNF-α for a further 15 min. Each time point required 4 × 107 neutrophils which were incubated in the presence of either 500 μg/ml MPO-ANCA or 500 μg/ml normal IgG at 37°C. At the appropriate time points, the reaction was stopped by rapid centrifugation (2500 r.p.m., 1 min), the supernatant discarded and the resulting cell pellet resuspended in 500 μl of ice cold extraction buffer (20 mm Tris (pH 7.4) 0.25 m sucrose, 10 mm DTT, 2 mm EDTA, 2 mm EGTA) with freshly added protease inhibitors (100 μg/ml aprotinin, 100 μg/ml pepstatin, 1 mm PMSF and 100 μg/ml leupetin) and immediately snap frozen in liquid nitrogen to lyse the neutrophils. The neutrophils were then further disrupted by sonication on ice. The resulting homogenates were centrifuged at 100 000 g for 45 min at 4°C, with the supernatant resulting from this spin (the cytosolic protein fraction) collected and maintained on ice. The pellet was resuspended in extraction buffer containing fresh protease inhibitors and 2% 3-[(cholamidopropyl)dimethylammonio]-1-propane-sulphate, left on ice for 5 min and sonicated on ice until the pellet had dispersed. Following centrifugation at 100 000 g for a further 45 min at 4°C the supernatant (the membrane protein fraction) was removed and kept on ice. A 15 μl aliquot of each of the protein fractions was removed for protein estimation, with the remainder resuspended in an equal volume of 2 × SDS loading buffer and boiled for 5 min. All extracts were stored at − 20°C until needed.
Anti-phosphotyrosine studies: cell stimulation and fractionation
Tyrosine phosphorylation was determined as described previously [25], with modifications. Neutrophils were resuspended to 5 × 106/ml in prewarmed PBSG, treated with 5 μg/ml cytochalasin B for 5 min and primed with 2 ng/ml TNF-α for a further 15 min. Primed neutrophil suspensions were divided into 1 ml aliquots and preincubated for 5 min at 37°C. Individual aliquots were stimulated with either 500 μg/ml ANCA IgG, 500 μg/ml normal IgG or 1 μm fMLP for the appropriate time. Reactions were stopped by rapid centrifugation for 30 s and the cellular pellet resuspended in ice cold 2 × SDS loading buffer. All samples were then boiled for 5 min to completely denature the proteins. Again, all fractions were stored at − 20°C. In some experiments, cytochalasin B and TNF-α primed neutrophils were incubated in the presence or absence of 150 μm genistein for 15–20 min prior to stimulation.
SDS-PAGE and immunoblotting
Neutrophil extracts were electrophoresed in 10% SDS- polyacrylamide gels. For the PKC studies, proteins were loaded at 20 μg per lane for the cytosolic proteins and 40 μg per lane for the membrane fractions. For the antiphosphotyrosine studies, neutrophil lysates were added at 1 × 106 cell equivalents. In all cases, proteins were transferred to nitrocellulose membranes. The resulting blots were blocked at room temperature for 1 h in 5% nonfat milk in TBST (10 mm Tris (pH 7.4), 150 mm NaCl and 0.05% Tween-20). For the antiphosphotyrosine studies, 2% BSA was substituted for the 5% nonfat milk at all stages. Blots were incubated at room temperature for at least 2 h with the appropriate antibodies diluted in TBST. This was followed by extensive washing of the membranes in TBST (five changes over 40 min). Subsequently, the blots were incubated for 1 h with an appropriate horseradish peroxidase-conjugated secondary antibody (dilution 1:2000) in 5% nonfat milk in TBST, washed thoroughly and visualized using enhanced chemiluminescence (Pierce).
Statistical analysis
All data are expressed as mean ± SEM. Where appropriate the results were analysed for level of significance using the Wilcoxon signed ranks test. P < 0.05 was considered to be statistically significant.
RESULTS
ANCA-induced superoxide production
In the absence of any priming event, isolated human neutrophils constitutively produced appreciable levels of O2− over the 60 min incubation period (4.69 ± 0.25 nmol), which remained largely unaffected by the addition of ANCA or normal IgG (Fig. 1a), with only fMLP producing a significant increase in O2− production (7.17 ± 0.66 nmol, P < 0.05).
Fig. 1.
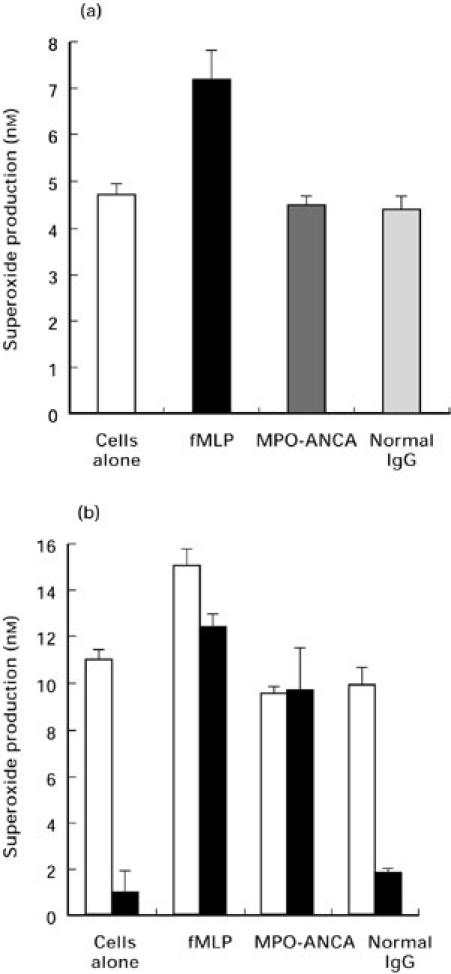
Effects of priming on O2− release from neutrophils. O2− production was measured in response to 100 μg/ml of either MPO-ANCA, or normal IgG and to 1 μm fMLP in the absence of priming (a) or in the presence 2 ng/ml TNF-α, alone (b, open bars) or in combination with 5 μg/ml cytochalasin B (b, solid bars). Results are expressed as nmol/1 × 105 cells/60 min. Data shown are mean ± SEM for at least two experiments performed in triplicate.
Priming with TNF-α alone increased the overall level of O2− produced (11.11 ± 0.47 nmol/100 min for cells alone), but did not permit stimulation with ANCA or normal IgG to cause a significant increase of O2− production above basal levels (Fig. 1b). Again, only stimulation with fMLP promoted a significant increase in O2− production (15.24 ± 0.69 nmol/100min, P < 0.05). It was only when neutrophils were pretreated with cytochalasin B (CTB) and primed with TNF-α that the ability of ANCA to stimulate increased levels of O2− was revealed. Figure 1(b) demonstrates that inclusion of CTB and TNF-α in the assay dramatically reduced the basal level of O2− production by cells alone (11.11 ± 0.47 nmol/100 min with TNF-α alone versus 1.0 ± 0.09 nmol/100 min with TNF-α and CTB), and in response to normal IgG (9.89 ± 0.82 nmol/100 min with TNF-α alone versus 1.76 ± 0.17 nmol/100 min with TNF-α and CTB). In contrast, considerable levels of O2− production were still produced in response to fMLP (15.24 ± 0.67 nmol/100 min with TNF-αversus 12.45 ± 0.57 nmol/100 min with TNF-α and CTB) or ANCA IgG (9.55 ± 0.33 nmol/100 min with TNF-α alone versus 9.66 ± 1.87 nmol/100 min with TNF-α and CTB).
The effects of ANCA on cytochalasin B treated, TNF-α primed human neutrophils were studied further. ANCA positive IgG lead to a time (Fig. 2) and dose (Fig. 3) dependent production of O2−. In contrast to the rapid production of O2− stimulated by fMLP, which was maximal within 10 min (Fig. 2), ANCA IgG stimulation showed an initial lag period of 10–15 min, where little or no O2− production was observed, with maximal production occurring at approximately 100 min. Normal IgG was non stimulatory. O2− production from MPO-ANCA stimulated neutrophils was consistently greater than from PR3-ANCA (Fig. 3). Thus, in all subsequent studies MPO-ANCA was added at 100 μg/ml and PR3-ANCA at 200 μg/ml to allow the study of comparable levels of O2− production from ANCA positive IgG.
Fig. 2.
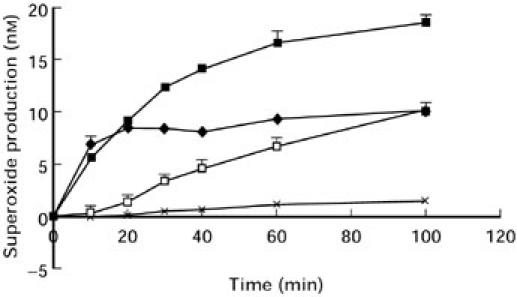
Time course of neutrophil O2− production (1 × 105 cells) in response to fMLP (♦), 100 ng/ml PMA (▪), 100 μg/ml ANCA IgG (□) or 100 μg/ml normal IgG (X). The results show mean ± SEM of a single experiment representative of at least three experiments performed in triplicate.
Fig. 3.
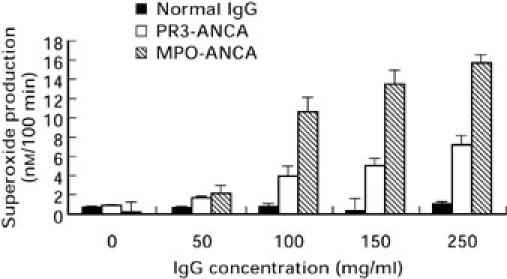
Effect of increasing IgG dose on neutrophil O2− production. Each value represents mean ± SEM of three experiments performed in triplicate
F(ab′)2 preparations of ANCA IgG failed to initiate O2− production (Fig. 4). Typical values for O2− production of whole antibody versus F(ab′)2 were; anti-PR3 ANCA 8.46 ± 2.25 versus 0.23 ± 0.35 nmol/100 min (P < 0.001), anti-MPO ANCA 11.67 ± 1.39 versus 1.48 ± 0.17 nmol/100 min (P < 0.05). The mean value for whole normal IgG was 1.39 ± 0.27 nmol/100 min.
Fig. 4.
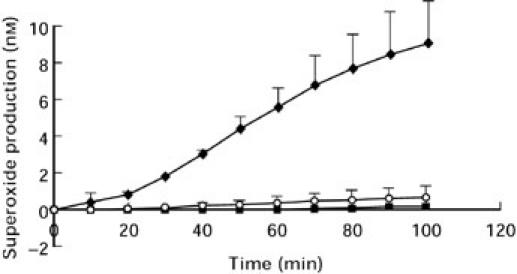
O2− production from neutrophils stimulated with 100 μg/ml MPO-ANCA IgG (♦) or 150 μg/ml of a corresponding F(ab′)2 fragment (○) or 100 μg/ml normal IgG (▪). Each point represents mean ± SEM of a single experiment performed in triplicate and is representative of at least three individual experiments.
Cross-linking
Neither pretreatment of neutrophils with ANCA F(ab′)2 (150 μg/ml) or subsequent cross-linking with 100 μg/ml of a sheep antihuman F(ab′)2 resulted in O2− release. In these experiments, neutrophil O2− production in response to ANCA F(ab′)2 was 1.06 ± 0.09 nmol/100 min/1 × 105 cells, in the presence of cross-linking antibody O2− production reached only 0.56 ± 0.12 nmol/100 min/1 × 106 cells. The cross-linking antibody alone had no effect on O2− production (0.80 ± 0.09 nmol/100 min/1 × 106 cells).
Effect of PKC inhibitors on ANCA-induced superoxide production
Initial studies using chelerythrine chloride suggested that PKC could be involved in ANCA induced O2− production. Thereafter, the potential role of PKC in the stimulation of O2− production by ANCA was examined using a range of PKC inhibitors chosen to distinguish between the involvement of those isoforms of PKC regulated by calcium (c-PKCs) and those that are calcium independent (n- and a-PKCs).
Chelerythrine chloride is a highly selective inhibitor of PKC, IC50 0.66 μm [22]. Figure 5 shows the dose responsive inhibition of O2− production observed when neutrophils were stimulated in the presence of increasing concentrations of chelerythrine chloride. Almost complete inhibition of the fMLP response (94.1 ± 1.4%) was observed at 50 μm chelerythrine chloride. Inhibition of the ANCA-induced response was highly significant at 74.7 ± 4.2% for PR3-ANCA and 72.6 ± 3.4% for MPO-ANCA. The PMA response (not shown) was inhibited by 90.3 ± 1.6% at 50 μm.
Fig. 5.
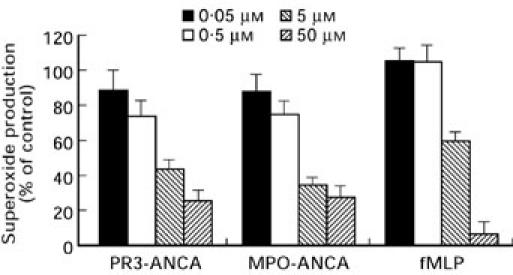
Effects of increasing concentrations of chelerythrine chloride (IC50 = 0.66 μm) on neutrophil O2− production stimulated by either, 200 μg/ml PR3-ANCA, 100 μg/ml MPO-ANCA or 1 μm fMLP. Results are expressed as percentage inhibition of the maximal effect seen with each stimulus. Data are shown as mean ± SEM of at least three individual experiments performed in triplicate.
The involvement of c-PKC isoforms was assessed using the selective c-PKC inhibitor, bisindolylmaleimide I. Whilst not being specific for the c-PKC isoforms (α, βI, βII and γ), bisindolylmaleimide I is over 10 times less potent as an inhibitor of the n-PKCs (δ and ε) and over 300 times weaker for the a-PKC ζ. Figure 6 demonstrates that bisindolylmaleimide I inhibited the response to fMLP by 41.0 ± 5.1% (P < 0.005). Inhibition of the ANCA induced O2− response was similar to that of fMLP, with 38.3 ± 9.8% for PR3-ANCA and 46.2 ± 8.6% for MPO-ANCA (P < 0.005 for both). Gö6976 is a potent PKC inhibitor with a high selectivity for the c-PKC isoforms and little or no ability to inhibit of the n- and a-PKCs. Inhibition of the ANCA induced O2− response (Fig. 7) was dose responsive and at the highest concentration of inhibitor used (50 nm) PR3-ANCA induced O2− production was inhibited by 63 ± 8.0% (P < 0.001) and MPO-ANCA by 57 ± 2.7% (P < 0.001), nearly as great as the inhibition seen with chelerytherine chloride. The effect of Gö6976 on the fMLP response was significant but less pronounced than that seen with ANCA, with inhibition reaching 30.4 ± 4.0% at 50 nm (P < 0.002).
Fig. 6.
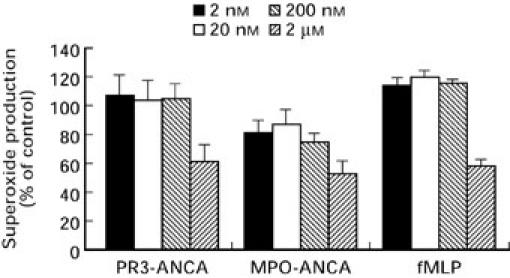
Effects of increasing concentrations of bisindolylmaleimide I (IC50 = 0.02 μm) on neutrophil O2− production stimulated by either, 200 μg/ml PR3-ANCA, 100 μg/ml MPO-ANCA or 1 μm fMLP. Results are expressed as percentage inhibition of the maximal effect seen with each stimulus. Data are shown as mean ± SEM of at least three individual experiments performed in triplicate.
Fig. 7.
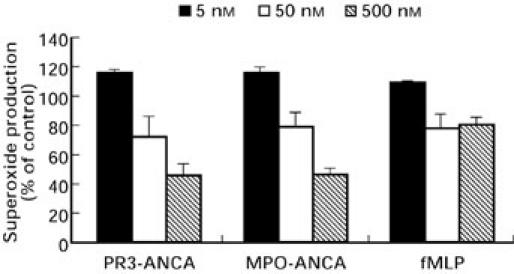
Effects of increasing concentrations of Gö6976 (IC50 = 5 nm) on neutrophil O2− production stimulated by either, 200 μg/ml PR3-ANCA, 100 μg/ml MPO-ANCA or 1 μm fMLP. Results are expressed as percentage inhibition of the maximal effect seen with each stimulus. Data are shown as mean ± SEM of at least three individual experiments performed in triplicate.
Effect of genistein on ANCA-induced superoxide production
Pretreatment of primed neutrophils with genistein led to a dose dependent reduction in O2− production (Fig. 8) stimulated by either ANCA, or fMLP. At 150 μm genistein the PR3-ANCA response was inhibited by 42.3 ± 6.3% (P < 0.001), the MPO-ANCA response by 44.5 ± 8.6% (P < 0.001) and the fMLP response by 56.1 ± 4.2% (P < 0.0001). No significant decrease in O2− production was seen following stimulation with PMA (8.6 ± 3.9%).
Fig. 8.
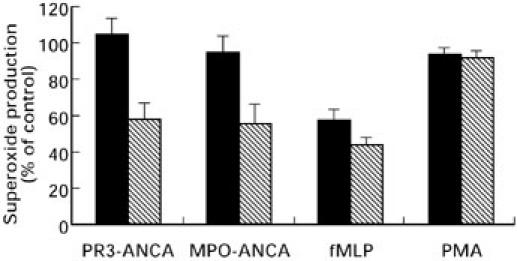
Effects of 25 μm (solid bar) and 150 μm (hatched bar) genistein on neutrophil O2− production stimulated by either, 200 μg/ml PR3-ANCA, 100 μg/ml MPO-ANCA, 1 μm fMLP or 100 ng/ml PMA. Results are expressed as percentage inhibition of the maximal effect seen with each stimulus. Data are shown as mean ± SEM of at least three individual experiments performed in triplicate.
Translocation/activation of PKC isoenzymes
PKC translocates from the cytosol to the membrane fraction of cells upon activation. Figure 9 shows the typical time-course of PKC translocation/activation in response to ANCA or normal IgG. It can be seen that for ANCA, but not normal IgG, PKCβII translocated from the cytosol to the membrane transiently at 5 and 10 min following stimulation. It can be seen that there was also a slight movement of PKCβII in response to normal IgG, but this appeared to occur later (10 min) and was not accompanied by an associated loss from the cytosol. The other PKC isoenzymes expressed in neutrophils; PKCα, PKCβI, PKCδ and PKCζ did not translocate in response to ANCA or normal IgG.
Fig. 9.

Translocation of PKC isoforms in response to ANCA or normal IgG. Cytosolic and membrane proteins were isolated from cytochalasin B and TNF-α primed neutrophils stimulated with either 500 μg/ml of ANCA IgG or 500 μg/ml normal IgG, with the reaction terminated at the time points indicated. The results were obtained using 20 μg of cytosolic protein or 40 μg/ml membrane protein for each lane.
Tyrosine phosphorylation of neutrophil proteins
Figure 10 demonstrates that the tyrosine phosphorylation of numerous neutrophil proteins was observed in response to ANCA, but not normal IgG. The typical time-course of tyrosine phosphorylation is also shown, and as it can be seen, increases in phosphorylation were transient, observed as early as 5 min, maximal at 10–15 min and almost completely absent by 20 min. Proteins phosphorylated in response to ANCA include those with molecular weights of; 34, 36, 38, 40, 42, 45, 74, 85, 110, 115 and 150 kDa. Figure 10 also demonstrates that pretreatment of the neutrophils with 150 μm genistein, completely abolished tyrosine phosphorylation in response to subsequent challenge with ANCA.
Fig. 10.

Time course of tyrosine phosphorylation in neutrophils stimulated with ANCA or normal IgG. Primed neutrophils (5 × 106/ml) were stimulated in the presence or absence of 500 μg/ml ANCA or normal IgG, or 500 μg/ml ANCA following pretreatment of the neutrophils with 150 μm genistein for 30 min. The reactions were terminated at the time points indicated. Results shown are representative of at least two other experiments.
DISCUSSION
Inappropriate neutrophil activation within the microvasculature is thought to play a significant role in the inflammation and tissue destruction seen in the systemic vasculitides. That ANCA are capable of neutrophil activation, with the release of lytic enzymes and reactive oxygen species, was first suggested by Falk et al. [4]. In addition, ANCA activation of neutrophils also results in the production of other pro-inflammatory agents such as IL-1β [20] and IL-8 [26], all of which serve to amplify the inflammatory focus. This diversity of responses must involve the recruitment and strict coordinated regulation of multiple signal transduction systems. The mechanism by which ANCA activates neutrophils has yet to be elucidated, although Fcγ receptor engagement and signal transduction processes dependent on PKC have been implicated [6,7,9,10].
The data presented here confirm that ANCA IgG are capable of inducing the activation of the neutrophil respiratory burst. Furthermore, our observations support a requirement for the intact IgG molecule and indicate that ANCA F(ab′)2 fragments, whilst being important for antigen recognition, do not provide a sufficient signal for full neutrophil activation. This is consistent with a reported role for Fcγ receptor IIa engagement in ANCA-induced O2− production [9,10,27]. There is controversy regarding the requirement of the Fc portion of ANCA IgG, even Kettritz et al. [8], whilst proposing that antigen recognition and binding of human ANCA F(ab′)2 was sufficient for neutrophil activation, found that antigen recognition by a mouse monoclonal PR3-ANCA F(ab′)2 was ineffective compared to the intact IgG molecule.
In agreement with other studies of ANCA-induced O2− production as assessed by the reduction of ferricytochrome C, our data demonstrate the requirement of pretreating neutrophils with cytochalasin B and TNF-α [4,8,10]. The kinetics of O2− production are different from that induced by the chemotactic peptide fMLP. In contrast to the rapid (< 5 min) production of O2− by fMLP, O2− production by ANCA shows an initial lag phase of 10–15 min, with maximal O2− production after 90 min. The time scale of the fMLP response demonstrates that the cellular machinery for O2− production is in place during the first 15 min. Thus, the lag seen with ANCA may be the result of the recruitment of specific signal transduction components unique for the ANCA induced response.
We show for the first time that protein kinase C and tyrosine kinases are involved in the mechanism by which ANCA activate neutrophils to produce reactive oxygen species (O2−). A role for PKC in ANCA activation of neutrophils has been suggested previously [6]. To our knowledge, this is the first report to show a role for a specific isoform of PKC, namely PKC βII in the activation of neutrophils by ANCA. Chelerythrine chloride is a highly selective and potent inhibitor of protein kinase C showing competitive inhibition with respect to substrate phosphorylation, but noncompetitive inhibition with respect to ATP [22]; thus the relatively high concentration of ATP found in most cells should not reduce the potency of chelerythrine in whole cell assays as used here. Whilst almost completely abolishing O2− production in response to either fMLP or PMA, chelerythrine inhibited the ANCA mediated response by a maximum of 70%. The effects of this highly specific PKC inhibitor indicate that PKC is involved in ANCA stimulated O2− production, although this is less dependent on PKC than the PMA or fMLP response in primed neutrophils.
Having established a role for PKC in ANCA induced O2− production, the involvement of the various groups of PKC isoforms was examined. Human neutrophils have been shown to possess members of the conventional and novel isoforms of PKC [14–16]. For this reason, PKC inhibitors were chosen to distinguish between these groups. The staurosporine analogue bisindolylmaleimide I shows an increased selectivity for the inhibition of Ca2+ sensitive c-PKC isoforms [23]. In the presence of bisindolylmaleimide I, O2− production in response to either fMLP or ANCA was decreased by a maximum of 45%. Whilst indicating that the c-PKCs play a role in the establishment of O2− production in response to ANCA, the lack of more extensive inhibition (compared to the 70% seen with chelerythrine) suggests that involvement of members of the other groups of PKC isoforms cannot be ruled out. To clarify the involvement of the c-PKC isoforms in the ANCA stimulated respiratory burst, Gö6976, a selective inhibitor of the c-PKC isoforms was used. The lack of extensive inhibition of the fMLP response by Gö6976 is in accordance with studies by others [28] and suggests that n-PKCs are required in addition to c-PKC isoforms to mediate maximal activation of the NADPH oxidase. The 60% inhibition seen with Gö6976 for the ANCA-induced response, strongly suggests a major role for c-PKC isoforms in ANCA activation of O2− production, with a smaller contribution from the n- (or possibly the a-) PKC isoforms.
The involvement of Ca2+-dependent isoforms in the ANCA response is further supported by the translocation of the βII isoform in response to ANCA (Fig. 10). This translocation was observed at times during, and towards the end of the lag period of O2− release. This strongly suggests that this isoform is involved in the establishment of the respiratory burst.
The production of O2− stimulated by ANCA was significantly reduced in the presence of the tyrosine kinase inhibitor genistein, suggesting that tyrosine phosphorylation plays a role in the establishment of the ANCA induced respiratory burst. However, the O2− response can only be partially dependent on tyrosine kinases as preincubation of the neutrophils with 150 μm genistein, while completely abolishing tyrosine phosphorylation in response to ANCA (Fig. 10), reduced O2− production by a maximum of 50% (Fig. 8). In accordance with previous reports genistein also significantly inhibited the fMLP response [29–31].
An increase in tyrosine phosphorylation of a number of neutrophil proteins has been observed in response to a variety of stimuli [32,33]. Here, it is demonstrated that ANCA activation of cytochalasin B and TNF-α primed neutrophils results in the tyrosine phosphorylation of numerous proteins in a time dependent manner. Increases in tyrosine phosphorylation were firmly established by 5 min post-stimulation and continued rising until 10–15 min when the response peaked and, in the majority of cases, was almost completely absent by 20 min. As with the recruitment of PKC βII, tyrosine phosphorylation peaks towards the end of the lag phase of O2− production, confirming that the lag phase is a period of active signal transduction.
In conclusion, intact ANCA IgG stimulated primed neutrophils to undergo a respiratory burst via mechanisms distinct from those used by fMLP or direct activation of PKC by PMA. The ANCA-induced response is heavily dependent on Ca2+-dependent isoforms of PKC, and requires the phosphorylation of tyrosine residues on a number of proteins. Furthermore, the inhibition of the ANCA stimulated respiratory burst by inhibitors of PKC provide an attractive basis for novel therapeutic strategies. Indeed, the anti-inflammatory effects of several PKC inhibitors have been demonstrated [34–37] and may provide one approach to more successful and specific therapies for patients with systemic vasculitis.
Acknowledgments
This work was supported by The Medical Research Council (MRC) of Great Britain. D.J.R. was in receipt of an MRC studentship. J.M.L. is a Royal Society University Research Fellow.
REFERENCES
- 1.Savage COS, Harper L, Adu D. Primary systemic vasculitis. Lancet. 1997;349:553–8. doi: 10.1016/s0140-6736(97)80118-3. [DOI] [PubMed] [Google Scholar]
- 2.Savage COS, Pottinger BE, Gaskin G, et al. Autoantibodies developing to myeloperoxidase and proteinase 3 in systemic vasculitis stimulate neutrophil cytotoxicity towards cultured endothelial cells. Am J Pathol. 1992;141:335–42. [PMC free article] [PubMed] [Google Scholar]
- 3.Ewert BH, Jennette JC, Falk RJ. Anti-myeloperoxidase antibodies stimulate neutrophils to damage human endothelial cells. Kidney Int. 1992;41:375–83. doi: 10.1038/ki.1992.52. [DOI] [PubMed] [Google Scholar]
- 4.Falk RJ, Terrell RS, Charles LA, et al. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charles LA, Falk RJ, Jennette JC. Reactivity of ANCA with mononuclear phagocytes. J Leukocyte Biol. 1992;51:65–8. doi: 10.1002/jlb.51.1.65. [DOI] [PubMed] [Google Scholar]
- 6.Lai KN, Lockwood CM. The effect of anti-neutrophil cytoplasm autoantibodies on the signal transduction in human neutrophils. Clin Exp Immunol. 1991;85:396–401. doi: 10.1111/j.1365-2249.1991.tb05738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai KN, Leung JCK, Rifkin I, et al. Effect of anti-neutrophil cytoplasm autoantibodies on the intracellular calcium concentration of human neutrophils. Lab Invest. 1994;70:152–62. [PubMed] [Google Scholar]
- 8.Kettritz R, Jennette JC, Falk RJ. Cross-linking of ANCA. antigens stimulates superoxide release by human neutrophils. J Am Soc Nephrol. 1997;8:386–94. doi: 10.1681/ASN.V83386. [DOI] [PubMed] [Google Scholar]
- 9.Porges AJ, Redecha PB, Kimberly WT, et al. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via FcγRIIa. J Immunol. 1994;153:1271–80. [PubMed] [Google Scholar]
- 10.Mulder AHL, Heeringa C, Brouwer E, et al. Activation of granulocytes by anti-neutrophil cytoplasmic antibodies (ANCA): an FcγRII- dependent process. Clin Exp Immunol. 1994;98:270–8. doi: 10.1111/j.1365-2249.1994.tb06137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hug H, Sarre TF. Protein kinase C isoenzymes. Divergence in signal transduction? Biochem J. 1993;291:329–43. doi: 10.1042/bj2910329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dekker LV, Parker PJ. Protein kinase C — a question of specificity. TIBS. 1994;19:73–7. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 13.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein-kinase-C. Science. 1992;258:607–14. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 14.Obel D, Rasmussen LH, Christiansen NO. Protein kinase C subtypes in human neutrophils. Scand J Clin Lab Invest. 1991;51:299–302. doi: 10.3109/00365519109091618. [DOI] [PubMed] [Google Scholar]
- 15.Majumdar S, Kane LH, Rossi MW, et al. Protein kinase C isotypes and signal-transduction in human neutrophils: selective substrate specificity of calcium-dependent β-PKC and novel calcium-independent nPKC. Biochim Biophis Acta. 1993;1176:276–86. doi: 10.1016/0167-4889(93)90056-u. [DOI] [PubMed] [Google Scholar]
- 16.Dang P, Hakim J, Perianin A. Immunochemical identification and transloaction of protein kinase C zeta in human neutrophils. FEBS Letts. 1994;349:338–42. doi: 10.1016/0014-5793(94)00700-4. [DOI] [PubMed] [Google Scholar]
- 17.Curnutte JT, Erickson RW, Ding J, et al. Reciprocal interactions between protein kinase C and components of the NADPH oxidase complex may regulate superoxide production by neutrophils stimulated with phorbol ester. J Biol Chem. 1994;269:10813–9. [PubMed] [Google Scholar]
- 18.Ptasznik A, Prossnitz ER, Yoshikawa D, et al. A tyrosine kinase signalling pathway accounts for the majority of phosphatidylinositol 3,4,5-trisphosphate formation in chemoattractant-stimulated human neutrophils. J Biol Chem. 1996;271:25204–7. doi: 10.1074/jbc.271.41.25204. [DOI] [PubMed] [Google Scholar]
- 19.Toothill VJ, Van-Mourik JA, Niewenhuis HK, et al. Characterization of the enhanced adhesion of neutrophil leukocytes to thrombin-stimulated endothelial cells. J Immunol. 1990;145:283–91. [PubMed] [Google Scholar]
- 20.Brooks CJ, King WJ, Radford DJ, et al. IL-1β production by human polymorphonuclear leukocytes stimulated by anti-neutrophil cytoplasmic autoantibodies: relevance to systemic vasculitis. Clin Exp Immunol. 1996;106:273–9. doi: 10.1046/j.1365-2249.1996.d01-835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayo LA, Curnutte JT. Kinetic microplate assay for superoxide production by neutrophils and other phagocytic cells. Methods Enzymol. 1990;186:567–75. doi: 10.1016/0076-6879(90)86151-k. [DOI] [PubMed] [Google Scholar]
- 22.Herbert JM, Augereau JM, Gleye J, et al. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Comm. 1990;172:993–9. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- 23.Toullec D, Pianetti P, Coste H, et al. The bisindolylmaleimide GF-109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–81. [PubMed] [Google Scholar]
- 24.Martiny-Baron G, Kazanietz MG, Mischak H, et al. Selective inhibition of protein kinase C isoenzymes by the indolocarbazole Go6976. J Biol Chem. 1993;268:9194–7. [PubMed] [Google Scholar]
- 25.Liang L, Huang C. Tyrosine phosphorylation induced by cross-linking of Fcγ–receptor type II in human neutrophils. Biochem J. 1995;306:489–95. doi: 10.1042/bj3060489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cockwell P, Brooks CJ, Adu D, et al. In vitro and in situ production of IL-8 in ANCA-associated vasculitis. Kidney Int. 1997;52:272–3. [Google Scholar]
- 27.Reumaux D, Vossebeld PJM, Roos D, et al. Effect of tumor necrosis factor-induced integrin activation on Fcγ receptor II-mediated signal transduction: relevance for activation of neutrophils by anti proteinase 3 or anti-myeloperoxidase antibodies. Blood. 1995;86:3189–95. [PubMed] [Google Scholar]
- 28.Wenzel-Seifert K, Schachtele C, Seifert R. N-protein kinase C isoenzymes may be involved in the regulation of various neutrophil functions. Biochem Biophys Res Comm. 1994;200:1536–43. doi: 10.1006/bbrc.1994.1625. [DOI] [PubMed] [Google Scholar]
- 29.Huang CK, Laramee GR, Casnellie JE. Chemotactic factor induced tyrosine phosphorylation of membrane associated proteins in rabbit peritoneal neutrophils. Biochem Biophys Res Comm. 1988;151:794–801. doi: 10.1016/s0006-291x(88)80351-6. [DOI] [PubMed] [Google Scholar]
- 30.Gomez-Cambronero J, Huang CK, Bonak VA, et al. Tyrosine phosphorylation in human neutrophil. Biochem Biophys Res Comm. 1989;162:1478–85. doi: 10.1016/0006-291x(89)90841-3. [DOI] [PubMed] [Google Scholar]
- 31.Naccache PH, Gilbert C, Caon AC, et al. Selective inhibition of human neutrophil functional responsiveness by erbstatin, an inhibitor of tyrosine protein-kinase. Blood. 1990;76:2098–104. [PubMed] [Google Scholar]
- 32.Kusunoki T, Higashi H, Hosoi S, et al. Tyrosine phosphorylation and its possible role in superoxide production by human neutrophils stimulated with FMLP and IgG. Biochem Biophys Res Commun. 1992;183:789–96. doi: 10.1016/0006-291x(92)90552-v. [DOI] [PubMed] [Google Scholar]
- 33.Rollet E, Caon AC, Roberge CJ, et al. Tyrosine phosphorylation in activated human neutrophils. Comparison of the effects of different classes of agonists and identification of the signaling pathways involved. J Immunol. 1994;153:353–63. [PubMed] [Google Scholar]
- 34.Cerna H, Fiala B, Han K, et al. Antiphlogistics in peridontistry. Acta Univ Palacki Olomuc Fac Med (Czechoslovakia) 1989;123:293–302. [PubMed] [Google Scholar]
- 35.Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunction in diabetic rats by an oral PKB β inhibitor. Science. 1996;272:728–31. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 36.Jacobson PB, Kuchera SL, Metz A, et al. Anti-inflammatory properties of Go 6850: a selective inhibitor of protein kinase C. J Pharm Exp Therapeut. 1995;275:995–1002. [PubMed] [Google Scholar]
- 37.Kuchera S, Barth H, Jacobson P, et al. Anti-inflammatory properties of the protein kinase C inhibitor3-[1-[3-(dimethylamino) propyl]-1H-indol-3-yl]-4-(1H-inol-3-yl)-1H-pyrrole-2,5-dione monochloride (GF109203X) in the PMA-mouse ear edema model. Agents Actions. 1993;39:C169–C73. doi: 10.1007/BF01972756. [DOI] [PubMed] [Google Scholar]