

Abstract
The various biological activities of butyrate have been well documented. In this study, we tested the effects of butyrate on TNF-α-induced complement C3 and factor B biosynthesis in human intestinal epithelial cells. The biosynthesis of C3, factor B and IL-8 was evaluated at the protein and mRNA levels. To evaluate transcriptional activation, the nuclear run-on assay was performed. The transcription factor–DNA binding activity was assessed by an electrophoretic gel mobility shift assay (EMSA). In the intestinal epithelial cell lines HT-29, T84 and Caco-2, sodium butyrate enhanced TNF-α-induced C3 secretion, but suppressed TNF-α-induced factor B and IL-8 secretion. Nuclear run-on assay revealed that transcriptional regulatory mechanisms are involved in the effects of sodium butyrate. The EMSAs indicated that sodium butyrate suppressed TNF-α-induced nuclear factor (NF)-κB– and activation protein (AP)-1–DNA binding activity, but enhanced TNF-α-induced activation of CCAAT/enhancer-binding protein (C/EBP)β (NF-IL-6)–DNA binding activity. Sodium butyrate induced a counter-regulatory effect on TNF-α-induced C3 and factor B biosynthesis in human intestinal epithelial cells. Butyrate action has been discussed with its activity to induce histone hyperacetylation, but its counter-regulatory effect on complement biosynthesis may be closely associated with the modulation of transcription factor activation.
Keywords: NF-κB, NF-IL-6, IL-8, transcription
INTRODUCTION
TNF-α is a potent immunomodulator derived from activated monocytes/macrophages and T lymphocytes [1]. TNF-α induces both systemic and local acute-phase responses, including the secretion of proinflammatory cytokines and complement proteins. The physiological importance of excess TNF-α is evident in patients with inflammatory bowel disease (IBD); TNF-α levels are elevated in the serum and stools of active IBD patients [2,3], and the number of TNF-α-secreting cells is locally increased in the diseased mucosa of IBD [4]. These findings indicate an important role for TNF-α-mediated immune responses in inflammatory conditions of the intestinal mucosa.
The complement system is a potent effector for normal immune and inflammatory responses [5,6], and there is increasing evidence to indicate local complement activation in the diseased mucosa of patients with IBD [7–9]. Complement activation occurs via three pathways: the classical, alternative and mannan-binding lectin (MBL)-initiated pathways [5,6]. Each pathway can result in the assembly of C3 cleaving enzyme complexes (C3 convertase) on the target cell surfaces. The C3 convertase of the alternative pathway, C3b,Bb, is a complex of C3 and factor B, and the generation of this complex is the rate-limiting step in the activation of the alternative pathway. In general, liver hepatocytes are regarded as the primary source of biosynthesis for most complement components, but recent reports have demonstrated the extrahepatic biosynthesis of complement proteins in a variety of tissues [10]. For example, in the human intestine, complement components are synthesized locally and secreted into the lumen [7], and we have recently found by in situ hybridization that transcripts of C3 and factor B are expressed in normal human colonic epithelial cells [11]. Proinflammatory cytokines such as IL-1β, IL-6 and TNF-α were shown to be potent inducers of C3 and factor B [10,12–14].
Dietary fibre (non-starch polysaccharides) and resistant starch escape digestion in the upper gastrointestinal tract and undergo anaerobic bacterial fermentation in the colon. This process produces short-chain fatty acids (SCFA) such as acetate, propionate and butyrate as the major by-product [15,16]. Of the most abundant SCFAs, butyrate has been shown to have significant effects on colonic epithelial cells both in vivo and in vitro. Butyrate serves as the primary energy source for the normal colonic epithelium [16] and stimulates the growth of the colonic mucosa [17], but in tumour cell lines it inhibits growth and induces apoptosis [18]. In some clinical studies, the effectiveness of butyrate enemas in the treatment of active ulcerative colitis has been reported [19–21]. The precise molecular mechanisms underlying this response have not been identified, but it is believed to be partially based on the inhibitory actions of butyrate on the production of proinflammatory mediators in the intestinal epithelium. For example, Gibson & Rosella have previously demonstrated an inhibitory action of butyrate on IL-8 secretion in isolated colonic crypt cells [22].
In this study, we demonstrate that sodium butyrate differentially modulates TNF-α-induced complement C3 and factor B biosynthesis in human intestinal epithelial cells. Especially, potent inhibitory effects on factor B biosynthesis, which may lead to a blockade of the activation of the alternative pathway, indicate the anti-inflammatory nature of sodium butyrate against complement-mediated inflammatory responses. One possible mechanism underlying counter-regulation by sodium butyrate may be mediated by rapid modulation of activation of transcription factors, including nuclear factor-κB (NF-κB) [23], CCAAT-enhancer binding protein (C/EBP)β (also known as NF-IL-6) [24] and activation protein (AP)-1 [25].
MATERIALS AND METHODS
Reagents
Recombinant human TNF-α (specific activity 2.5 × 106 U/mg by cytotoxic assay against LM cells) was kindly provided by Dainippon Pharmaceutical Co. (Osaka, Japan). All other reagents used in this study were purchased from Sigma Chemical Co. (St Louis, MO).
Cells
Human colonic adenocarcinoma cell lines, HT-29, Caco-2 and T84 [26], were obtained from the American Type Culture Collection (ATCC; Rockville, MD). HT-29 cells were maintained in RPMI 1640 (Gibco Labs, Grand Island, NY) containing 10% fetal bovine serum (FBS; Gibco), and Caco-2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 20% FBS. T84 cells were cultured in a 1:1 mixture of DMEM and Ham's F-12 medium (Gibco) containing 10% FBS. All culture media were supplemented with 50 U/ml penicillin and 50 μg/ml streptomycin. All experiments were performed after reaching confluence.
Quantification of human C3 and factor B
The levels of antigenic C3 and factor B in the samples were determined by sandwich ELISA established in our laboratory [13,14]. The lower limit of detection was 1 ng/ml of C3 and factor B. Levels of human IL-8 were detected using ELISA kit purchased from Bio-Source (Camarillo, CA).
Northern blot analysis
Total cellular RNA was isolated by the acid guanidinium thiocyanate-phenol-chloroform method [27], and Northern blotting was performed according to the methods described previously [28,29]. The cDNA probes were prepared as follows: two cDNA fragments of 2.4 and 1.8 kb specific for human C3 were isolated by ClaI/SalI double digestion of the human C3 cDNA clone pHLC3.11 (ATCC) [30]. A 2.9-kb empty plasmid vector (pAT153/Pvu) was also isolated. Likewise, 1.6-kb and 0.7-kb ClaI/BamHI restriction fragments of the human factor B cDNA clone pFB3b (kindly provided by Dr R. D. Campbell, Oxford, UK) [31] were used as hybridization probes. A human IL-8 cDNA probe was generated from monolayer of human umbilical vein endothelial cells using reverse transcription-polymerase chain reaction (RT-PCR), and the following oligonucleotide (5′ ACATGACTTCCAAGCTGGCC 3′) corresponding to nucleotide position 101–121 of the sequence published by Matsushima et al. [32] as a sense primer, and oligonucleotide (3′ TTTTATGA ATTCTCAGCCCT 5′) corresponding to nucleotide position 404–385 as anti-sense primer, as described previously [28]. The 300-base pair PCR products were subcloned into TA cloning vector (Promega, Madison, WI) and sequenced by dideoxynucleotide chain termination method [33]. The human β-actin clone HFBCA46 was purchased from ATCC.
Nuclear run-on assays
Nuclei from confluent HT-29 monolayers were performed essentially as described [34]. Briefly, cells were exposed to TNF-α (50 ng/ml) or TNF-α (50 ng/ml) plus sodium butyrate (5 mm) for 3 h, scraped off and lysed by buffer (10 mm Tris pH 7.4, 10 mm NaC1, 3 mm MgC12, 0.5% Nonidet P-40). After centrifugation the nuclei were resuspended in storage buffer (50 mm Tris pH 8.3, 40% glycerol, 5 mm MgCl2, 0.1 mm EDTA), and stored at −80°C. Run-on transcription was performed by each 5.0 × 106 nuclei in 200 μl transcription buffer (20% glycerol, 50 mm Tris pH 8.0, 0.1 mm EDTA, 5 mm MgCl2, 0.5 mm DTT, 500 μm each of adenosine triphosphate, cytidene triphosphate, and guanosine triphosphate, and 200 μCi α-32P-uridine triphosphate). Subsequently, nuclear RNA was extracted by the acid guanidinium thiocyanate-phenol-chloroform method [27], and hybridized to denatured, filter-bound cDNA probes for 48 h. Hybridization and washing procedures were performed according to the methods described above.
Nuclear extracts and electrophoretic gel mobility shift assays
Nuclear extracts were prepared from HT-29 cells exposed to TNF-α (50 ng/ml), sodium butyrate (5 mm), and TNF-α (50 ng/ml) plus sodium butyrate (5 mm) for 1.5 h by the method of Dignam et al. [35]. Consensus oligonucleotides of NF-κB (5′ AGT TGA GGG GAC TTT CCC AGC C) [23], AP-1 (c-jun: 5′ CGC TTG ATGAGT CAG CCG GAA) [25], C/EBP (TGC AGA TTG CGC AAT CTG CA) [24], and Sp1 (5′ ATT CGA TCG GGG CGG GGC GAG) [36] were used. The consensus sequences for each of the transcription factors are underlined. Oligonucleotides were 5′ end-labelled with T4 polynucleotide kinase (Promega) and γ-32P-ATP (Amersham). Binding reactions were performed by preincubating 7.5 μg of nuclear proteins in HEPES (20 mm; pH 7.9), KCl (60 mm), MgCl2 (1 mm), EDTA (0.1 m), glycerol (10%), dithiothreitol (0.5 mm), and poly (dI-dC; 2 μg) on ice for 10 min, followed by addition of the 32P-labelled oligonucleotide and a second incubation at room temperature for 20 min. Samples were loaded directly on non-denaturing 4% polyacrylamide gels prepared in Tris-glycin-EDTA buffer pH 8.5. The gels were dried and exposed to Kodak XRP film with intensifying screen. Supershift experiments were performed as described above except that 1 μl of antibody to each transcription factor was added to the binding mixture in the absence of the labelled probe. Antisera specifically recognizing each transcriptional factor were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Experiments with unlabelled oligonucleotides used a 100-fold molar excess relative to the radiolabelled oligonucleotide.
Densitometry of hybridization signals
The intensity of each band was analysed by the program NIH Image version 1.58 after scanning by Image Scanner EPSON GT-9000 (Tokyo, Japan).
RESULTS
Effects of sodium butyrate on the secretion of complement C3, factor B and IL-8 in HT-29 cells
HT-29 cells were incubated for 24 h with medium alone, TNF-α 25 ng/ml, or TNF-α 25 ng/ml plus increasing concentrations of sodium butyrate. As shown in Fig. 1a, TNF-α induced C3 secretion, and sodium butyrate dose-dependently enhanced this TNF-α-induced C3 secretion. As demonstrated in Fig. 1b, TNF-α also induced a marked increase in factor B secretion. However, in contrast to the effects on C3 secretion, sodium butyrate inhibited TNF-α-induced factor B secretion in a dose-dependent fashion. These effects of sodium butyrate were observed at concentrations as low as 0.5 mm and reached a plateau at a concentration of 2.5 mm. The presence of sodium butyrate (5 mm) enhanced TNF-α-induced C3 secretion by 5.6-fold, but suppressed TNF-α-induced factor B secretion by 72%. To characterize further the effect of sodium butyrate on TNF-α-induced responses, we tested the effect of sodium butyrate on IL-8 secretion in HT-29 cells (Fig. 1c). TNF-α has been established as a potent inducer of IL-8 secretion in HT-29 cells [37]. TNF-α induced a marked increase in IL-8 secretion, but this was also dose-dependently suppressed by sodium butyrate. In these experiments the addition of sodium butyrate alone had no effect. Similar responses in C3 and factor B secretion were also detected in other intestinal epithelial cell lines T84 and Caco-2 (Fig. 2).
Fig. 1.
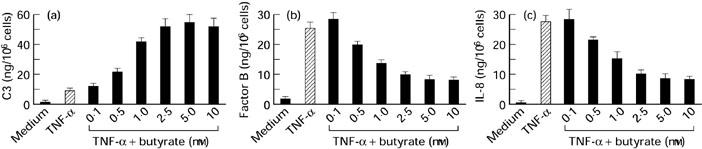
Effects of sodium butyrate on TNF-α-induced C3 (a), factor B (b) and IL-8 (c) secretion in HT-29 cells. Cells were incubated with medium alone, TNF-α (25 ng/ml), or TNF-α (25 ng/ml) plus increasing concentrations of sodium butyrate for 24 h. The amounts of C3, factor B and IL-8 were determined by ELISA. Values were expressed as mean ± s.d. of quadruplicate cultures.
Fig. 2.
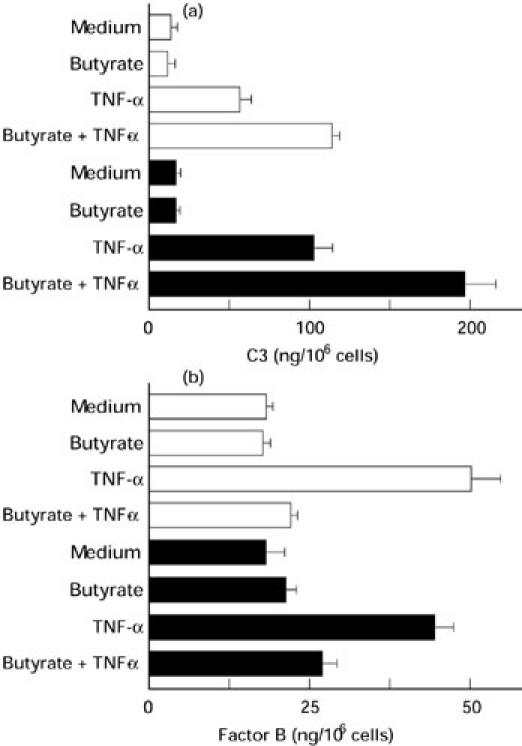
Effects of sodium butyrate on TNF-α-induced C3 (a) and factor B (b) secretion in T84 and Caco-2 cells. Cells were incubated with medium alone, sodium butyrate 5 mm, TNF-α 25 ng/ml, or sodium butyrate 5 mm plus TNF-α 25 ng/ml for 24 h. The amounts of C3 and factor B were determined by ELISA. Values were expressed as mean ± s.d. of quadruplicate cultures. □, T84; ▪, Caco-2.
Sodium butyrate modulates C3, factor B and IL-8 mRNA abundance
We examined the effects of sodium butyrate on the abundance of C3, factor B and IL-8 mRNA in HT-29 cells (Fig. 3). The cells were cultured for 6 h with medium alone, sodium butyrate (5 mm), TNF-α (25 ng/ml) or sodium butyrate (5 mm) plus TNF-α (25 ng/ml). Northern blot analysis demonstrated that unstimulated HT-29 cells weakly expressed the factor B gene only. The addition of TNF-α induced a marked increase in the abundance of C3 (5.2 kb), factor B (2.4 kb) and IL-8 (1.8 kb) mRNA. The addition of sodium butyrate alone exerted no effect, but enhanced TNF-α-induced C3 mRNA abundance by 2.5-fold and inhibited TNF-α-induced factor B and IL-8 mRNA abundance by 75% and 70%, respectively.
Fig. 3.
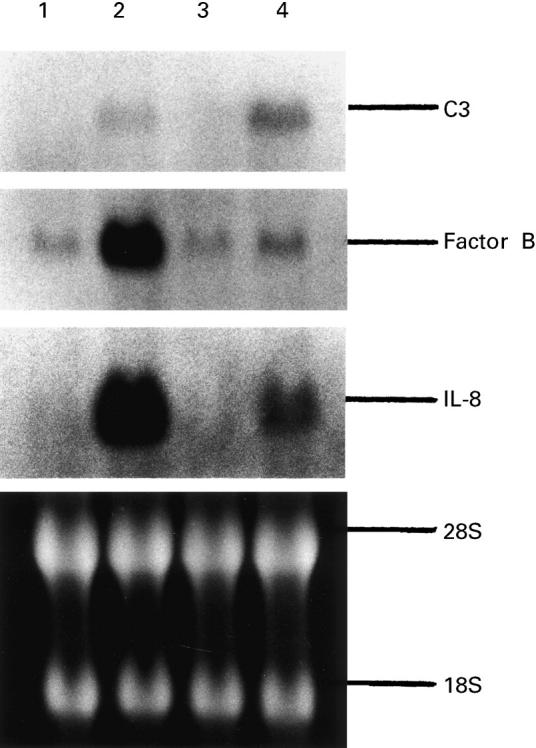
Northern blot analysis of C3, factor B and IL-8 mRNA expression in HT-29 cells. Cells were incubated with medium alone, TNF-α 25 ng/ml, sodium butyrate 5 mm, or TNF-α 25 ng/ml plus sodium butyrate 5 mm for 6 h, and then total cellular RNA was extracted. Lane 1, medium alone; lane 2, TNF-α; lane 3, sodium butyrate 5 mm; lane 4, TNF-α 25 ng/ml plus sodium butyrate 5 mm.
Sodium butyrate differentially modulates transcriptional rate of the C3, factor B and IL-8 genes
To investigate further how sodium butyrate modulated C3, factor B and IL-8 mRNA abundance in HT-29 cells, we performed nuclear run-on assays to evaluate the changes in transcriptional activation of these genes. As shown in Fig. 4, stimulation with TNF-α for 3 h induced a marked increase in the transcriptional rate of the C3, factor B and IL-8 genes (three-fold in C3, four-fold in factor B, and 20-fold in IL-8). Sodium butyrate enhanced TNF-α-induced transcriptional activation of the C3 gene by 2.5-fold, but decreased TNF-α-induced transcriptional activation of the IL-8 and factor B genes by 65% and 55%, respectively. Similar results were observed in two other experiments. These results indicate that transcriptional regulatory mechanisms are involved in the effects of sodium butyrate on C3, factor B and IL-8 mRNA expression. The empty plasmid (pAT153/PvuII) indicates weak non-specific bindings.
Fig. 4.
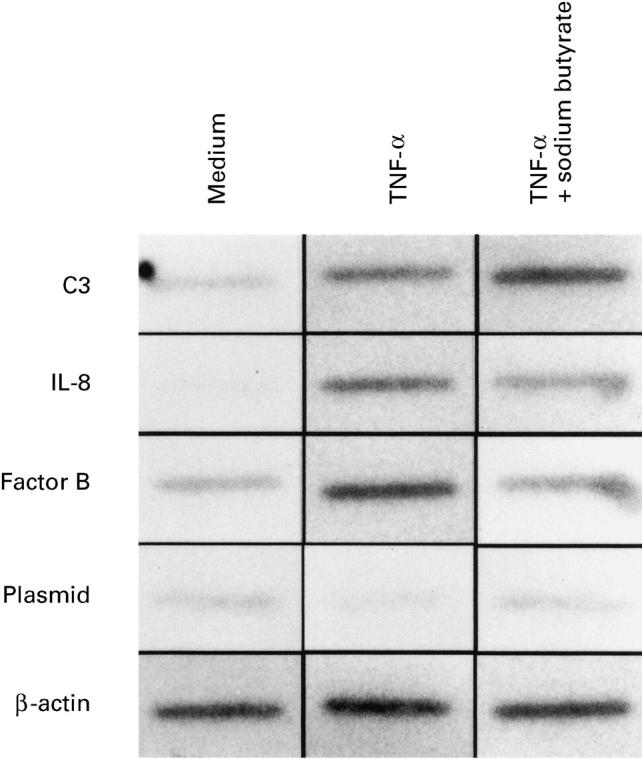
Nuclear run-on assays of C3, IL-8 and factor B gene transcription in HT-29 cells. Confluent HT-29 monolayers were stimulated with TNF-α 25 ng/ml or TNF-α 25 ng/ml plus sodium butyrate 5 mm for 3 h, and then nuclei were prepared. Run-on transcription was performed in 5.0 × 106 nuclei in the presence of α-32P-uridine triphosphate for 60 min, and nuclear RNA was isolated and hybridized to filter-bound DNA probe. Plasmid, a 2.9-kb empty plasmid (pAT153/Pvu) isolated from human C3 cDNA clone pHLC3.11.
Modulation of transcription factor activation by sodium butyrate
The importance of transcription factor activation in the regulation of many genes has been reported [23–25,35,36]. In particular, the expression of several genes during acute-phase responses, including C3, factor B and IL-8 biosynthesis, is regulated by the activation of specific transcription factors such as NF-κB, C/EBPβ (NF-IL-6) and AP-1 [23–25,35,36,38,39]. To elucidate the mechanisms underlying the transcriptional responses to sodium butyrate, we evaluated the activation of the transcription factors such as NF-κB, C/EBPβ, AP-1 and SP-1 in HT-29 cells. As demonstrated in Fig. 5a, stimulation with TNF-α for 1.5 h induced a marked increase in NF-κB–DNA binding activity (lane 3), and the specificity of this reaction was confirmed by the addition of cold oligo-DNA where the reactive band disappeared (lane 5). Interestingly, the addition of sodium butyrate potently suppressed the TNF-α-induced activation of NF-κB binding activity (70% inhibition, lane 4). The addition of the antibodies against a 50 000 mol. wt subunit (p50) of NF-κB and a 65 000 mol. wt subunit (p65) induced supershifts of the binding complexes (lanes 6 and 7), indicating that this binding complex was a heterodimer consisting of p50 and p65 subunits [23]. In Fig. 5b, AP-1 (c-jun) binding activity is shown; AP-1–DNA binding activity was observed in unstimulated HT-29 cells, and TNF-α enhanced this AP-1–DNA binding activity. The addition of sodium butyrate also suppressed the TNF-α-induced AP-1–DNA binding activity (35% inhibition, lane 4).
Fig. 5.
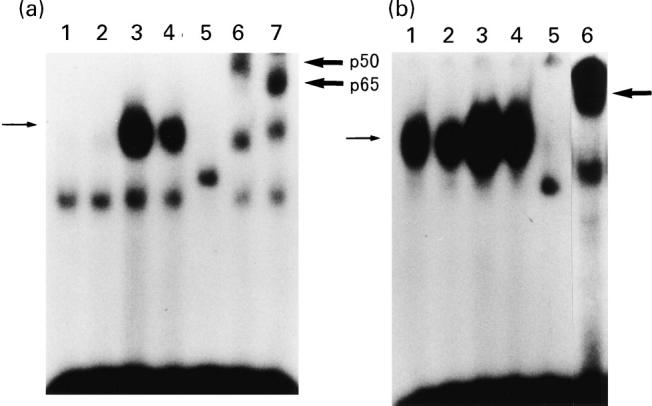
Electrophoretic gel mobility shift assays (EMSAs) for NF-κB (a) and AP-1 (b) DNA binding activities. HT-29 cells were incubate with medium alone, sodium butyrate 5 mm, TNF-α 25 ng/ml, or sodium butyrate 5 mm plus TNF-α 25 ng/ml for 1.5 h, and then nuclear extracts were prepared. (a) Lane 1, medium alone; lane 2, sodium butyrate; lane 3, TNF-α; lane 4, sodium butyrate plus TNF-α; lane 5, TNF-α plus cold probe; lane 6, TNF-α plus anti-p50 antibody; lane 7, TNF-α plus anti-p65 antibody. (b) Lane 1, medium alone; lane 2, sodium butyrate; lane 3, TNF-α; lane 4, sodium butyrate plus TNF-α; lane 5, TNF-α plus cold probe; lane 6, TNF-α plus anti-c-jun antibody. Thin arrow on the left indicates specific band, and thick arrow on the right indicates supershift band.
Next, we tested the effects of sodium butyrate on TNF-α-induced C/EBP–DNA binding activity (Fig. 6a). C/EBP–DNA binding activity can be already observed in unstimulated HT-29 cells (lane 1), and this was markedly enhanced by the addition of TNF-α (lane 3). The specificity of this binding was also demonstrated by the addition of cold probe (lane 5). This binding complex was supershifted by the addition of anti-C/EBPβ (NF-IL-6; lane 7) [34], but was not altered by anti-C/EBP α and δ (lanes 6 and 8) [40]. This indicated that TNF-α specifically increased the C/EBPβ (NF-IL-6)-DNA binding activity. In contrast to the responses observed for NF-κB and AP-1, sodium butyrate enhanced C/EBPβ (NF-IL-6)-DNA binding activity (lane 4). Densitometric analysis indicated that sodium butyrate enhanced TNF-α-induced C/EBPβ–DNA binding by two-fold. We further tested the effects of sodium butyrate on Sp1–DNA binding activity. Sp1 was initially identified as a HeLa cell-derived factor that selectively activates transcription from the SV40 promoter [36]. As demonstrated in Fig. 6b, Sp1 binding activity was not affected by the addition of either TNF-α or sodium butyrate. These observations indicate that the effects of both TNF-α and sodium butyrate were specific for NF-κB, C/EBPβ (NF-IL-6) and AP-1 activation.
Fig. 6.
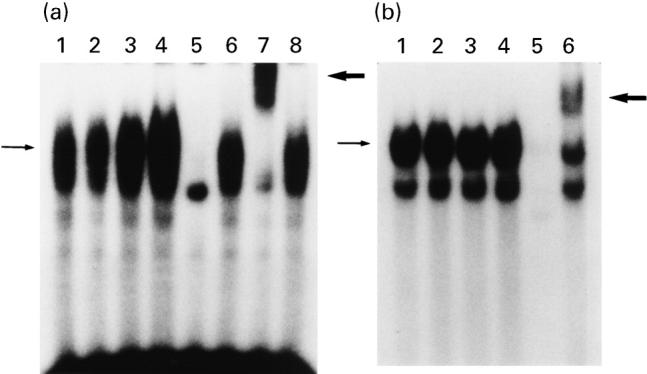
Electrophoretic gel mobility shift assays (EMSAs) for C/EBP (a) and Sp1 (b) DNA binding activities. HT-29 cells were incubate with medium alone, sodium butyrate 5 mm, TNF-α 25 ng/ml, or sodium butyrate 5 mm plus TNF-α 25 ng/ml for 1.5 h, and then nuclear extracts were prepared. (a) Lane 1, medium alone; lane 2, sodium butyrate; lane 3, TNF-α; lane 4, sodium butyrate plus TNF-α; lane 5, TNF-α plus cold probe; lane 6, TNF-α plus anti-C/EBP α antibody; lane 7, TNF-α plus anti-C/EBPβ (NF-IL-6) antibody; lane 8, TNF-α plus anti-C/EBP δ antibody. (b) Lane 1, medium alone; lane 2, sodium butyrate; lane 3, TNF-α; lane 4, sodium butyrate plus TNF-α; lane 5, TNF-α plus cold probe; lane 6, TNF-α sample plus anti-Sp1 antibody. Thin arrow on the left indicates specific band, and thick arrow on the right indicates supershift band.
DISCUSSION
To the best of our knowledge, this study is the first to demonstrate that the TNF-α-induced biosynthesis of complement C3 and factor B in intestinal epithelial cells is differentially regulated by sodium butyrate. It is of particular interest to note that the molecular events underlying these responses were mediated by different mechanisms between the C3 and factor B (and IL-8) genes. The major mechanism whereby sodium butyrate modulates the expression of several gene has been reported to depend on its ability to induce histone hyperacetylation [18,41–43]. However, it is likely that the regulation of the specific activation of the transcription factors NF-κB, NF-IL-6 and AP-1 DNA binding activity may be associated with the actions of sodium butyrate.
In HT-29 cells, sodium butyrate dose-dependently enhanced TNF-α-induced C3 secretion, but inhibited TNF-α-induced factor B and IL-8 secretion. Sodium butyrate induced similar responses in C3, factor B and IL-8 mRNA expression. These responses with respect to C3 and factor B secretion were also observed in T84 and Caco-2 cells. In the intestinal tract, the induction of complement C3 and factor B is beneficial for the host tissues as an enhancement of the local immunological defence activity [5]. However, this response simultaneously enhances alternative pathway-mediated cytotoxicity to the epithelial cells [28,29], since complement activation releases many biologically active peptides and can directly attack the epithelial cells [5]. In this study, we could not determine whether the overall effect of sodium butyrate on C3 and factor B secretion is anti-inflammatory or not. However, it may be possible to consider that the potent inhibitory effect of sodium butyrate on TNF-α-induced factor B biosynthesis leads to a blockade of alternative pathway activation at the initial steps of complement cascade. To clarify this point, further examination to evaluate the complement activity in supernatants will be required.
There is a discrepancy in the TNF-α-induced up-regulation of factor B between the run-ons and the protein/mRNA results. Although the transcriptional activation of factor B gene was detected as four-fold, both protein and mRNA expression agreed with a value over 10-fold. Such differences were not observed in IL-8 and C3 secretion. This indicates that transcriptional activation is not the sole mechanism for TNF-α-induced up-regulation of factor B gene. It has previously been reported that a post-transcriptional mechanism such as modulation of mRNA stability plays an important role in the interferon-gamma (IFN-γ)-induced up-regulation of factor B gene [44,45]. It is likely that similar post-transcriptional mechanisms may be involved in the TNF-α-induced up-regulation of factor B gene.
It remains unclear how sodium butyrate alters inflammatory stimuli-induced transcription factor–DNA binding activity. It is well known that the activation of specific transcription factors such as NF-κB, C/EBP-like protein and AP-1 is required to induce the transcription of a wide variety of genes that respond to immune or inflammatory signals. Indeed, consistent with the presence of NF-κB and C/EBP binding sites in the promotor region of the human C3 and factor B genes [46–48], both NF-κB and C/EBPβ (NF-IL06) can drive the expression of these genes. In addition to these factors, the AP-1 binding site is also required for IL-8 gene transcription in most cell types [49]. In order to clarify the molecular mechanisms underlying the mode of action of sodium butyrate in the regulation of C3, factor B and IL-8 gene activation, we performed electrophoretic gel mobility shift assays (EMSAs) using oligonucleotide probes for the transcription factors, NF-κB, C/EBPβ (NF-IL-6), and AP-1 (c-jun). Furthermore, since some of the actions of sodium butyrate are mediated via the activation of transcription factor Sp1–DNA binding activity [50], we tested Sp1 activation in our model. Our results showed that the NF-κB p50/p65 heterodimer DNA-binding complex was rapidly induced by stimulation with TNF-α, but the addition of sodium butyrate potently attenuated this response. TNF-α increased AP-1–DNA binding activity, but sodium butyrate also suppressed this response. In contrast to these phenomena, sodium butyrate clearly enhanced TNF-α-induced C/EBPβ (NF-IL-6)–DNA binding activity. Since Sp1–DNA binding activity was not affected by either TNF-α or sodium butyrate, TNF-α and sodium butyrate were specific for NF-κB, C/EBPβ (NF-IL-6) and AP-1 (c-jun).
In previous studies, the roles of NF-κB, C/EBPβ (NF-IL-6) and AP-1 have been well characterized for the IL-8 gene transcription [38,48–50]. NF-κB activation is the most critical step for IL-8 gene transcription, and C/EBPβ (NF-IL-6) and AP-1 activation are also required for TNF-α-induced IL-8 transcriptional activation. Co-operation between NF-κB and C/EBPβ (NF-IL-6) or AP-1 is sufficient for IL-8 gene activation, and C/EBPβ (NF-IL-6) is regarded as the primary co-operator. These findings suggest that the mechanisms by which sodium butyrate inhibited TNF-α-induced IL-8 gene expression may be mainly associated with the inhibition of NF-κB–DNA binding activity. The mild suppressive effects on AP-1–DNA binding activity might also participate in this process. Since the importance of NF-κB activation has also been characterized for factor B gene transcription, a similar mechanism might participate in the inhibitory effect of sodium butyrate on factor B gene expression. In addition, it will be necessary to determine how sodium butyrate affects the post-transcriptional regulation of factor B mRNA expression in the future. On the other hand, it has been reported that the induction of the human C3 gene was mainly mediated by the activation of C/EBPβ (NF-IL-6) [29]. Although the importance of C/EBP δ activation in C3 gene transcription has been reported in an IL-1β-stimulated hepatoma cell line [40], we could not detect such an activation of C/EBP δ in TNF-α-treated HT-29 cells. Therefore, we concluded that the sodium butyrate-induced activation of C/EBPβ (NF-IL-6) was the main mechanism by which sodium butyrate enhanced C3 gene expression. Thus, sodium butyrate differentially and specifically modulates the activation of various transcription factors which regulate the expression of inflammatory response genes, and the equilibrium between the altered activation of these transcription factors may determine part of the cellular responses to inflammatory stimuli.
It is generally accepted that butyrate leads to the development of reversible histone hyperacetylation via the inhibition of histone deacetylase activity [41–43]. Reversible histone hyperacetylation is now believed to play an important role in the regulation of the chromatin structure. Histone hyperacetylation leads to a more relaxed chromatin structure, and thus leads to facilitating transcription factor access to the promoter regions of certain genes, without directly initiating gene transcription [43]. Based on this knowledge, the mechanisms by which sodium butyrate induces the marked increase in TNF-α-induced C3 gene expression can be explained by at least two general mechanisms. The first involves an enhancement of transcriptional potential by histone hyperacetylation; however, this might not directly increase C3 gene transcription. The other mechanism involves an enhancement of the TNF-α-induced activation of C/EBPβ (NF-IL-6), which directly promotes C3 gene transcription. A combination of these two mechanisms might produce the up-regulation of TNF-α-induced C3 gene expression mediated by sodium butyrate. On the other hand, increased transcriptional potential by histone hyperacetylation might not account for the inhibitory effect on factor B and IL-8 secretion. Although transcriptional potential was increased by sodium butyrate, a suppression of NF-κB (and/or AP-1) DNA binding activity resulted in the inhibitory effect of sodium butyrate on factor B and IL-8 gene expression. It is now well recognized that the activation of the NF-κB system is closely associated with the inhibitory actions of the IκB system [51]. In order to clarify the mechanisms by which sodium butyrate suppresses NF-κB–DNA binding activity, the interactions between sodium butyrate and the activation of the IκB system must be determined.
In conclusion, this study demonstrated the counter-regulatory effects of sodium butyrate, a normal metabolite of bacteria, on the secretion of complement C3 and factor B in intestinal epithelial cells. The molecular mechanisms by which sodium butyrate regulates complement gene expression in intestinal epithelial cells may be dependent on both histone hyperacetylation and the modulation of transcription factor activation.
Acknowledgments
This study was supported in part by Grant-in-Aid for Scientific Research from the Ministry of Education, Science, and Culture, Japan (9470135 and 9670541).
REFERENCES
- 1.Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol. 1992:411–52. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- 2.Murch SH, Lamkin VA, Savagr MO, et al. Serum concentrations of tumor necrosis factor-α in childhood chronic inflammatory bowel disease. Gut. 1991;32:913–6. doi: 10.1136/gut.32.8.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braegger CP, Nicholls SW, Murch SH, et al. Tumor necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet. 1992;339:89–91. doi: 10.1016/0140-6736(92)90999-j. [DOI] [PubMed] [Google Scholar]
- 4.Breese EJ, Michte CA, Nicholls SW, et al. Tumor necrosis factor α-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology. 1994;106:1455–66. doi: 10.1016/0016-5085(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 5.Morgan BP. The complement system. In: Morgan BP, editor. Complement: clinical aspects and relevance to disease. London: Academic Press; 1990. pp. 1–35. [Google Scholar]
- 6.Thiel S, Vorup-Jensen T, Strover CM, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997;386:506–10. doi: 10.1038/386506a0. [DOI] [PubMed] [Google Scholar]
- 7.Ahrenstedt O, Knutson L, Nilsson B, et al. Enhanced local production of complement components in the small intestines of patients with Crohn's disease. N Engl J Med. 1990;322:1345–9. doi: 10.1056/NEJM199005103221903. [DOI] [PubMed] [Google Scholar]
- 8.Halstensen TS, Mollnes TE, Brandtzaeg P. Persistent complement activation in submucosal blood vessels of active inflammatory bowel disease: immunohistochemical evidence. Gastroenterology. 1989;97:10–19. doi: 10.1016/0016-5085(89)91409-1. [DOI] [PubMed] [Google Scholar]
- 9.Halstensen TS, Mollness TE, Garred P, et al. Epithelial deposition of immunoglobulin G1 and activated complement (C3b and terminal complement complex) in ulcerative colitis. Gastroenterology. 1990;98:1264–71. doi: 10.1016/0016-5085(90)90343-y. [DOI] [PubMed] [Google Scholar]
- 10.Colten HR, Strunk RC. Synthesis of complement components in liver and at extrahepatic sites. In: Whaley K, Loos M, Weiler JM, editors. Complement in health and disease. Dordrecht. Kluwer: Academic Publishers; 1993. pp. 127–58. [Google Scholar]
- 11.Andoh A, Fujiyama Y, Sakumoto H, et al. Detection of complement C3 and factor B gene expression in normal colorectal mucosa, adenomas and carcinomas. Clin Exp Immunol. 1998;111:477–83. doi: 10.1046/j.1365-2249.1998.00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molmenti EP, Ziambaras T, Perlmutter DH. Evidence for an acute phase response in human intestinal epithelial cells. J Biol Chem. 1993;268:14116–24. [PubMed] [Google Scholar]
- 13.Andoh A, Fujiyama Y, Bamba T, et al. Differential cytokine regulation of complement C3, C4, and factor B synthesis in human intestinal epithelial cell line, Caco-2. J Immunol. 1993;151:4239–47. [PubMed] [Google Scholar]
- 14.Andoh A, Fujiyama Y, Sumiyoshi K, et al. Modulation of complement C3, C4, and factor B production in human intestinal epithelial cells: differential effects of TNF-α, IFN-γ, and IL-4. Pathophysiology. 1995;2:251–9. [Google Scholar]
- 15.Cummings JH. Short chain fatty acids in the human colon. Gut. 1981;22:763–79. doi: 10.1136/gut.22.9.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. 1980;21:793–8. doi: 10.1136/gut.21.9.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheppach W, Bortram P, Richter F, et al. The effect of short-chain fatty acids on the human colonic mucosa in vitro. J Parent Enter Nutr. 1992;16:43–48. doi: 10.1177/014860719201600143. [DOI] [PubMed] [Google Scholar]
- 18.McBain JA, Eastman A, Nobel CS, et al. Apoptotic death in adenocarcinoma cell lines induced by butyrate and other histone deacerylase inhibitors. Biochem Pharmacol. 1997;53:1357–68. doi: 10.1016/s0006-2952(96)00904-5. [DOI] [PubMed] [Google Scholar]
- 19.Breuer RI, Buto SK, Christ ML, et al. Rectal irrigation with short chain fatty acids for distal ulcerative colitis. Dig Dis Sci. 1991;36:185–7. doi: 10.1007/BF01300754. [DOI] [PubMed] [Google Scholar]
- 20.Scheppach W, Sommer H, Kirchner T, et al. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology. 1992;103:51–56. doi: 10.1016/0016-5085(92)91094-k. [DOI] [PubMed] [Google Scholar]
- 21.Steihart AH, Brzezinski A, Baker JP. Treatment of refractory ulcerative proctosigmoiditis with butyrate enemas. Am J Gastroenterol. 1994;89:179–83. [PubMed] [Google Scholar]
- 22.Gibson P, Rosella O. Interleukin 8 secretion by colonic crypt cells in vitro: response to injury suppressed by butyrate and enhanced in inflammatory bowel disease. Gut. 1995;37:536–43. doi: 10.1136/gut.37.4.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lenardo MJ, Baltimore D. NF-κB: pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–9. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 24.Akira S, Isshiki H, Sugita T, et al. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990;9:1897–906. doi: 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bohmann D, Bos TJ, Admon A, et al. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcriptional factor AP-1. Science. 1987;238:1386–92. doi: 10.1126/science.2825349. [DOI] [PubMed] [Google Scholar]
- 26.Zweibaum A, Laburthe M, Grasset E, et al. Use of cultured cell lines in studies of intestinal cell differentiation and function. In: Schultz SG, Field RA, Frizzell RA, Rauner BB, editors. Handbook of physiology: the gastrointestinal system IV. Bethesda: American Physiological Society; 1989. pp. 223–55. [Google Scholar]
- 27.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 28.Andoh A, Fujiyama Y, Sumiyoshi K, et al. Interleukin-4 acts as an inducer of decay-accelerating factor gene expression in human intestinal epithelial cells. Gastroenterology. 1996;111:911–8. doi: 10.1016/s0016-5085(96)70058-6. [DOI] [PubMed] [Google Scholar]
- 29.Andoh A, Fujiyama Y, Sumiyoshi K, et al. Tumor necrosis factor-α up-regulates decay-accelerating factor gene expression in human intestinal epithelial cells. Immunology. 1997;90:358–63. doi: 10.1111/j.1365-2567.1997.00358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Bruijn MHL, Fey GH. Human complement component C3: cDNA coding sequence and derived primary structure. Proc Natl Acad Sci USA. 1985;82:708–12. doi: 10.1073/pnas.82.3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morley BJ, Campbell RD. Internal homologies of the Ba fragment from human complement component factor B, a class III MHC antigen. EMBO J. 1984;3:153–7. doi: 10.1002/j.1460-2075.1984.tb01776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsushima K, Morishita K, Yoshimura T, et al. Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MDNCF) and the induction of MDCF mRNA by interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167:1883–93. doi: 10.1084/jem.167.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–7. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts S, Bently DL. Distinct modes of transcription read through or termination at the c-myc attenuator. EMBO J. 1992;11:1085–93. doi: 10.1002/j.1460-2075.1992.tb05147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dignam JP, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucl Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dynan WS, Tjian R. Isolation of transcription factors that discriminate between different promoters recognized by RNA polymerase II. Cell. 1983;32:669–80. doi: 10.1016/0092-8674(83)90053-3. [DOI] [PubMed] [Google Scholar]
- 37.Eckmann L, Jung HC, Schurer-Maly C, et al. Differential cytokine expression by human intestinal epithelial cell line: regulated expression of interleukin 8. Gastroenterology. 1993;105:1689–97. doi: 10.1016/0016-5085(93)91064-o. [DOI] [PubMed] [Google Scholar]
- 38.Kunsh C, Lang RK, Rosen CA, et al. Synergistic transcriptional activation of the IL-8 gene by NF-kappa B p65 (RelA) and NF-IL6. J Immunol. 1994;153:153–64. [PubMed] [Google Scholar]
- 39.Stein B, Baldwin AS. Distinct mechanisms for regulation of the interleukin-8 gene involve synergism and cooperativity between C/EBP and NF kappa B. Mol Cell Biol. 1993;13:7191–8. doi: 10.1128/mcb.13.11.7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Juan TSC, Wilson DR, Wilde MD, et al. Participation of the transcription factor C/EBP δ in the acute-phase regulation of the human gene for complement component C3. Proc Natl Acad Sci USA. 1993;90:2584–8. doi: 10.1073/pnas.90.7.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kruh J, Defer N, Trichonicky L. Butyrate and the molecular biology of the large bowel. In: Rhombeau JL, Cummings JH, Sakata T, editors. Short chain fatty acids: metabolism and clinical importance. Columbus: Ross Laboratories; 1991. pp. 45–50. [Google Scholar]
- 42.Sealy L, Chalky R. The effect of sodium butyrate on histone modification. Cell. 1978;14:115–21. doi: 10.1016/0092-8674(78)90306-9. [DOI] [PubMed] [Google Scholar]
- 43.Arts J, Lansink M, Grimbergen J, et al. Stimulation of tissue-type plasminogen activator gene expression by sodium butyrate and trichostatin A in human endothelial cells involves histone acetylation. Biochem J. 1995;310:171–6. doi: 10.1042/bj3100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lappin DF, Guc D, Hill A, et al. Effect of interferon-γ on complement gene expression in different cell types. Biochem J. 1992;281:437–42. doi: 10.1042/bj2810437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchel TJ, Naughton M, Norsworthy P, et al. IFN-γ up-regulates expression of the complement components C3 and C4 by stabilization of mRNA. J Immunol. 1996;156:4429–34. [PubMed] [Google Scholar]
- 46.Wilson DR, Juan TSC, Wilde MD, et al. A 58-base-pair region of the human C3 gene confers synergistic inducibility by interleukin-1 and interleukin-6. Mol Cell Biol. 1990;10:6181–91. doi: 10.1128/mcb.10.12.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu L, Morley BJ, Campbell RD. Cell-specific expression of the human complement protein factor B gene: evidence for the role of two distinct 5′-flanking elements. Cell. 1987;30:331–42. doi: 10.1016/0092-8674(87)90436-3. [DOI] [PubMed] [Google Scholar]
- 48.Nonaka M, Gitlin JD, Colten HR. Regulation of human and murine complement: comparison of 5′ structural and functional elements regulating human and murine complement factor B gene expression. Mol Cell Biol. 1989;89:1–14. doi: 10.1007/BF00228274. [DOI] [PubMed] [Google Scholar]
- 49.Yasumoto K, Okamoto S, Mukaida N, et al. Tumor necrosis factor α and interferon γ synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-κB-like binding sites of the interleukin 8 gene. J Biol Chem. 1992;267:22506–11. [PubMed] [Google Scholar]
- 50.Nakano K, Mizuno T, Sowa Y, et al. Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line. J Biol Chem. 1997;272:22199–206. doi: 10.1074/jbc.272.35.22199. [DOI] [PubMed] [Google Scholar]
- 51.Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]