

Abstract
SLE is an autoimmune disease characterized by a wide range of anti-cellular and anti-nuclear autoantibodies. Many of these antigens are exposed or altered during apoptosis when the nucleus is dismantled in a controlled manner by caspases. We used Western blotting techniques to demonstrate that autoantibodies in SLE sera recognize antigens released during apoptosis. Reproducible bands, not seen in the untreated cells, with the characteristics of histones were seen when staining apoptotic cell lysates with SLE sera. Normal sera recognized some of these bands but much less strongly. Different triggers of apoptosis did not produce marked differences in the antigens recognized. We also compared different cell lines (Jurkat and U937) and found that the staining differed for one autoantigen in particular. The differential release of autoantigens by apoptotic cells may have relevance to the variety of autoantibodies seen in SLE.
Keywords: systemic lupus erythematosus, apoptosis, histones
INTRODUCTION
SLE is an autoimmune disease characterized by antibodies to a wide range of cellular and nuclear autoantigens, including DNA, ribonucleoproteins (Ro, La), poly ADP ribose polymerase (PARP), nucleosomes and histones [1]. Many of these antigens are exposed or altered during apoptosis, which is an active process leading to the controlled death of cells. Initial membrane changes are followed by the dismantling of the nucleus and other intracellular organelles. Some of the dismantled components are packaged and form on the surface of these apoptotic cells as ‘blebs’. Casciola-Rosen et al. showed by immunofluorescence that antibodies in sera from SLE patients were able to recognize antigens contained within these blebs and hypothesized that apoptosis is an important source of antigen in SLE [2]. They and others have demonstrated that the caspase enzymes involved in the destruction of the nucleus cleave certain well-known autoantigens (e.g. DNA-protein kinase, PARP, U1-RNP) revealing potentially autoreactive epitopes [3–5]. An important characteristic of apoptosis is the cleavage of chromatin into nucleosomal units by an endonuclease (caspase-activated DNase (CAD)) which is activated by caspases [6]. On a DNA gel these nucleosomes produce a ‘ladder’ of oligomers composed of different numbers of nucleosomes [6]. Nucleosomes are composed of DNA wound around a histone core, which comprises four histone subunits, i.e. a tetramer of H3 and H4 make up the central unit, attached either side of which are dimers of H2A and H2B. Externally bound H1 molecules hold adjacent nucleosomes together.
Autoantibodies to histones are frequently associated with SLE (reviewed in [7,8]). These include antibodies reactive with free histones, but also antibodies to histone–DNA complexes and whole nucleosomes: in particular, anti-H2AH2B-DNA antibodies are common in idiopathic SLE and in drug-induced lupus (reviewed in [9]). Histone-specific T cells have also been isolated from SLE patients [10,11].
Using gel electrophoresis and Western blotting techniques, we investigated the reactivity of SLE and normal sera with soluble proteins derived from apoptotic and healthy cells. We induced apoptosis by a variety of methods and used different cell lines. The different methods of inducing apoptosis showed few differences in the pattern of bands seen. We consistently found three to five distinct bands with the characteristics of histones. However, the different cell lines used showed a difference in one band in particular.
MATERIALS AND METHODS
Cell lines
Jurkat (human T cell lymphoma) and U937 (human monocyte-like histiocytic lymphoma) cell lines were grown in suspension. They were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 10 mm HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mml-glutamine (Life Technologies, Paisley, UK) at 37°C in 5% CO2. The cultures were maintained at 3–9 × 105 cells/ml, and dead cells were removed using Histopaque-1077 (Sigma, Poole, UK) 24 h prior to inducing apoptosis.
Induction of apoptosis
Time course and dose–response experiments were carried out to determine the optimum yield of apoptotic cells with a minimum increase in necrotic cells. Cyclohexamide (Sigma) was dissolved in methanol and diluted in PBS prior to adding to the cells (1 × 106/ml); the optimum conditions were 100 μg/ml for 3 h at 37°C, and this gave 70–80% apoptotic cells. Methanol diluted in PBS alone did not induce apoptosis. With UV light, 10 min exposure of cells in a 75-cm3 flask (1 × 106 cells/ml), with a low setting of the transilluminator (UVP) followed by 3 h incubation at 37°C gave 80–90% apoptotic cells. Anti-CD95 MoAb (DX2 clone, Pharmingen; Becton Dickinson, Oxford, UK) was used at 1 μg/ml for 18 h at 37°C with 1 × 105 cells/ml; this gave 20–40% apoptotic cells. Mouse IgG (Sigma) was used as an isotype control and no apoptosis was induced by it.
Detection of apoptosis
Annexin V-FITC and propidium iodide (PI) were used to distinguish apoptotic, viable and necrotic cells by flow cytometry. Annexin V binds to phosphatidylserine in the plasma membrane that becomes exposed during apoptosis and necrosis, PI is only able to enter cells with permeable membranes (necrotic) to bind with DNA. Cells (2 × 105) were stained using Annexin V-FITC kit (BenderMed Systems; Biowhittaker Ltd, Wokingham, UK): they were washed in PBS and resuspended in 200 μl binding buffer with 3 μl of Annexin V-FITC and were incubated for 10 min room temperature, washed in PBS and resuspended in 200 μl binding buffer again. PI was added (5 μl) and the cells were acquired by FACS (Becton Dickinson, Oxford, UK) immediately. The data were analysed using the WinMDI program. The induction of apoptosis was also confirmed morphologically. The characteristic blebbing and nuclear condensation is shown in Fig. 1.
Fig. 1.
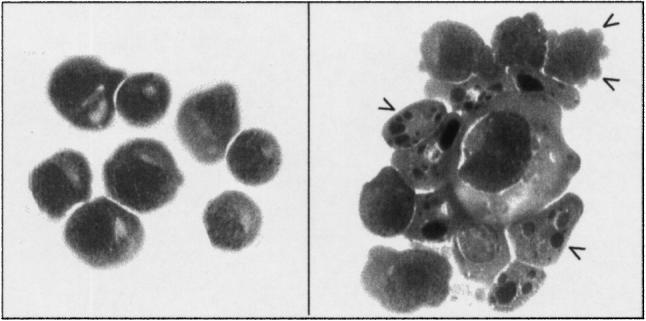
Morphological changes that occur during the induction of apoptosis in Jurkat cells after treatment with cyclohexamide. Left panel, untreated cells, right panel apoptotic cells. Arrows indicate blebs, and nuclear condensation and fragmentation. Cytospins were stained with haematoxylin and eosin.
Gel electrophoresis and protein blotting
Apoptotic and untreated cells (20 × 106 each) were washed with PBS and the pellets were resuspended in 200 μl lysis buffer (20 mm Tris–HCl/20 mm NaOH, 1 μg/ml aprotinin, 2 mm PMSF, 0.2% Triton X-100 (Sigma)) and incubated for 1 h at 4°C, mixing every 10–15 min. The lysate was spun at 10 000 g for 10 min at 4°C to pellet insoluble material and the supernatant was aspirated and placed on ice. A bicinchoninic acid (BCA) protein assay (Sigma) was performed to calculate the protein concentration in the samples, they were diluted in lysis buffer at 2 mg/ml and frozen at −70°C until required. For electrophoresis, the lysates (2 mg/ml) were mixed 1:1 with tricine sample buffer containing 2.5% mercaptoethanol (Novex, Frankfurt, Germany) and heated at 95°C for 3 min, then spun for 1 min at 10 000 g. Purified histones (1 mg/ml) were also treated with sample buffer as above. The histone used was total histone (Roche, Lewes, UK). The samples (20 μl/lane for 10-lane gels or 100 μl/lane on two-well gels) were loaded on 10–20% Tricine–SDS minigels (Novex), 10 μl of colour marker (Sigma) were also used and any blank wells contained sample buffer. The gel was run for 1.5–2 h at 125 V in tricine–SDS running buffer. The electrophoresed proteins were then transferred to nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany) using a semidry blotter unit for 3 h at 0.8 mA/cm (Sigma). The nitrocellulose was blocked overnight at 4°C in 5% milk powder (Marvel, Premier Beverages, Adbaston, UK) in PBS.
Western blotting
The blotted cellular proteins were probed with human antisera and a mouse anti-histone antibody (Roche). Sera from SLE patients were taken during routine clinics: all the patients met at least four American College of Rheumatology criteria. Normal sera were from healthy volunteers.
Either the blocked nitrocellulose membranes were cut into 3-mm strips from the centre of each lane, or an Immunotec Miniblotter was used on the whole membrane. The blocking solution was aspirated and the membranes were incubated with antibodies for 3 h at 37°C. The patient and normal sera were diluted 1:100 in PBS–2% bovine serum albumin (BSA); mouse anti-histone antibody (Roche) was used at 5 μg/ml. The strips were washed in PBS–0.1% Tween. Alkaline phosphatase-conjugated secondary antibodies were diluted in PBS–BSA (anti-human IgG (1:5000) for sera and anti-mouse IgG (1:1000) for anti-histone antibody (Sigma)) and incubated with the membranes for 1 h at 37°C. They were washed with PBS–Tween again and BCIP-NBT substrate (one tablet in 10 ml dH2O; Sigma) was added for 30–60 min until bands had developed. The strips were finally washed with PBS–Tween and dried on blotting paper.
Absorption of antibodies with antigen
To determine whether the binding of antibodies with the bands on the Western blots was specific, the antibodies were preabsorbed with antigen. SLE or normal sera were mixed 1:1 with histone (1 mg/ml) (Roche) to give a final serum dilution of 1:100. They were incubated overnight at 4°C prior to use in staining Western blots.
RESULTS
Antibody detection of proteins released from apoptotic Jurkat cells
The soluble lysate fraction of untreated Jurkat cells (i.e. not induced to undergo apoptosis) gave numerous bands on Western blots when probed with SLE serum IgG antibodies (Fig. 2), whereas very few if any bands were obtained when blots were similarly probed with normal sera (Figs 2 and 3). This is consistent with the well characterized occurrence of IgG autoantibodies to constitutive cellular autoantigens in SLE. These are likely to include autoantibodies to Ro, La and U1-RNP. The occurrence of the latter was demonstrated using a human serum containing antibodies specific for U1-RNP (Binding Site Ltd, Birmingham, UK): this was seen as a 70-kD band in the untreated cells (Fig. 4). Comparison of individual SLE sera showed both similarities and differences in the banding patterns they produced, indicating the occurrence of autoantibodies with both shared and individual specificities. For example, a strong band at approx. 35–38 kD and a doublet at approx. 19 kD (indicated by asterisks in Fig. 2b) were detected by SLE sera 21, 39 and 67, but not by SLE sera 4 and 34. All of these bands are likely to represent cytoplasmic autoantigens since the mild lysis conditions used on the cells (0.2% Triton X-100) would not solubilize nuclear material, which would therefore be present in the pellet rather than the supernatant following centrifugation of the lysates (see Materials and Methods).
Fig. 2.
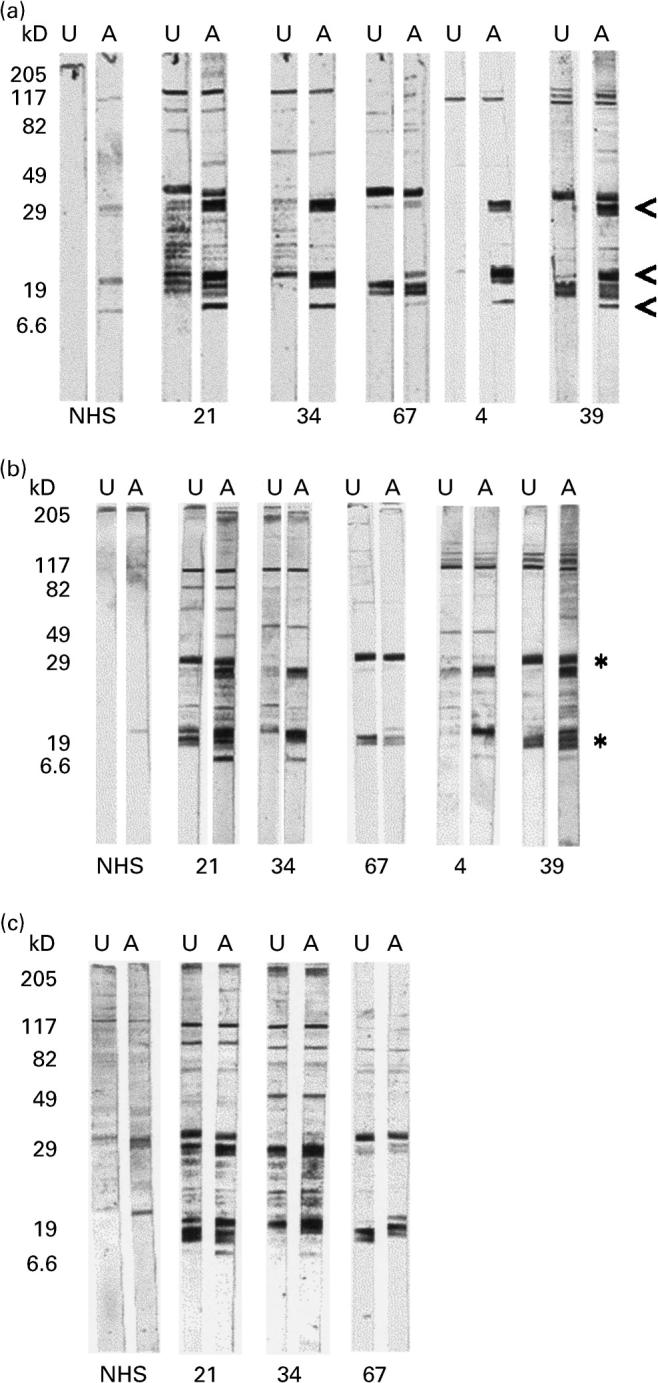
Western blot of apoptotic (A) and untreated (U) Jurkat cell lysates stained with SLE sera and normal human serum (NHS). Apoptosis was induced by (a) UV irradiation, (b) cyclohexamide and (c) anti-Fas antibody. Arrows indicate prominent bands in apoptotic cell preparations. *Bands present in both apoptotic and untreated preparations.
Fig. 3.
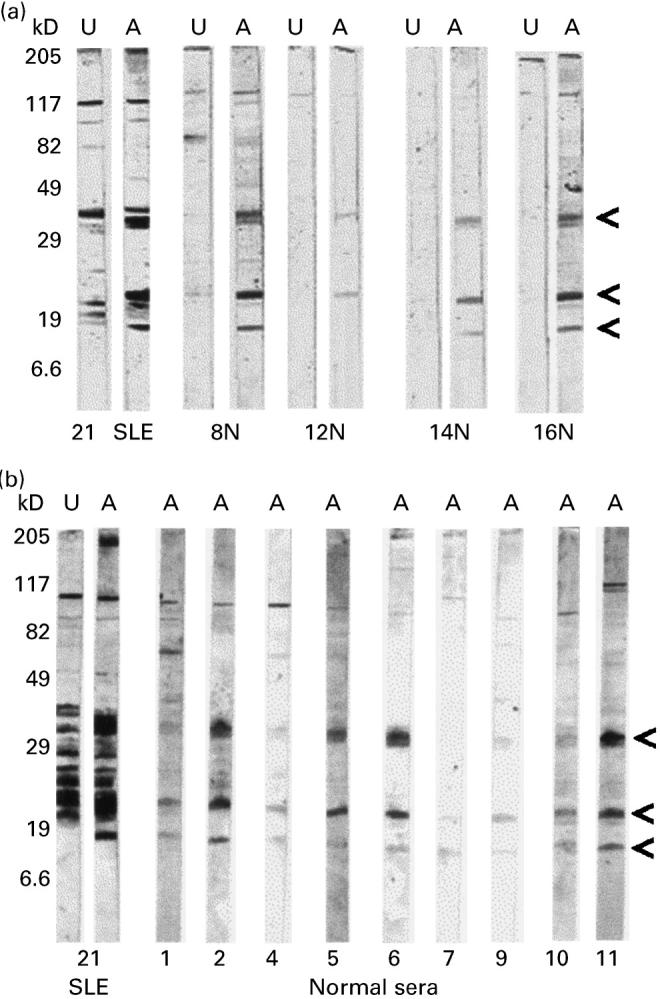
Western blot of UV-irradiated Jurkat cell lysates stained with normal sera (or SLE serum 21). Arrows indicate prominent bands in the apoptotic cell lysates (A) not seen in the untreated cell (U) lysates.
Fig. 4.
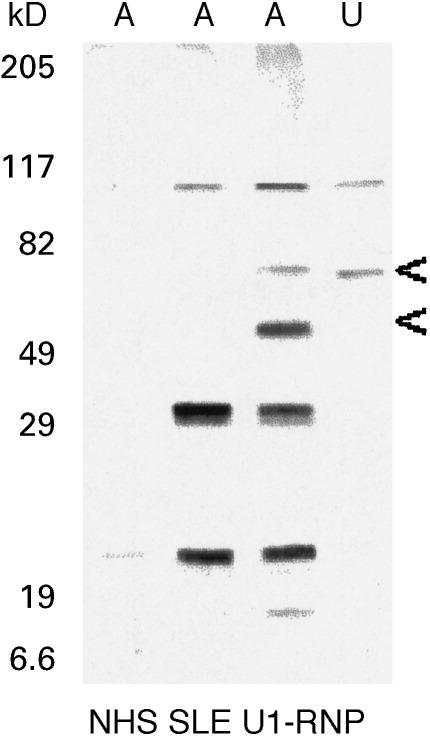
Western blot of cyclohexamide-treated (A) and untreated (U) Jurkat cell lysates, stained with normal human serum (NHS), SLE patient's serum and commercial human serum known to contain antibodies to U1-RNP. Arrows indicate U1-RNP-specific bands.
Similar Western blot probing of lysates of apoptotic Jurkat cells with SLE serum IgG antibodies revealed additional bands which were either not present, or were much weaker, in the Western blots of untreated Jurkat cells (Fig. 2). This suggests that additional proteins recognized by IgG autoantibodies are released into the soluble lysate fraction as a result of apoptosis, and possibly that some autoantigenic proteins are cleaved during apoptosis to yield new bands on the Western blots. The additional autoantigens detected are likely to include nuclear components due to the breakdown of the nucleus and chromatin during apoptosis.
Some of the additional bands obtained with apoptotic Jurkat lysates were revealed by only certain SLE sera. However, three particularly prominent bands were recognized in apoptotic (but not untreated) Jurkat lysates by IgG autoantibodies in most of the SLE sera (indicated by the arrows in Fig. 2a). These proteins migrated with approximate apparent molecular weights of 33 kD, 21 kD and 13 kD. They were detected by all five SLE sera employed, being strongly stained using SLE sera 21, 34, 4 and 39, but weakly stained with SLE serum 67. Also, detection of the 13-kD band was variable. The same three bands were also detected by IgG antibodies in most normal human sera employed, although much more weakly than by the patients' sera (Figs 2 and 3).
The banding patterns obtained with the SLE sera on Western blots of the apoptotic Jurkat lysates were similar whether apoptosis was induced by UV radiation (Fig. 2a), cyclohexamide (Fig. 2b) or anti-Fas MoAb (Fig. 2c). The bands were slightly weaker for the anti-Fas antibody-treated cells, because the percentage of apoptotic cells in the lysate was lower (20–40%) than for UV- or cyclohexamide-treated cells (70–90%).
Identification of the prominent antigenic bands in apoptotic Jurkat lysates as histones
The three prominent bands consistently detected on Western blots of the soluble lysate fraction of apoptotic Jurkat cells gave a pattern with the position and intensity expected for histones [8,9] (Fig. 5). Thus, the 33-kD band (which often appears as a doublet) coincides with the position of histones H1a and H1b; the broad band at 21 kD is consistent with histones H3, H2b and H2a which migrate very close together; the band at 13 kD is in the position for histone H4, although a cleavage product of H3 produced in apoptotic cells has been reported to migrate at a very similar position [12]. The staining of histone bands was confirmed by the following experiments. First, SLE IgG serum antibodies gave similar patterns of staining on Western blots of both apoptotic Jurkat cell lysates and purified histones (Fig. 6a). These same staining patterns were also given by mouse anti-histone antiserum (Fig. 6a). Second, the staining of both histones and apoptotic Jurkat proteins by SLE IgG serum antibodies was greatly reduced by preincubating the SLE serum with purified histones (Fig. 6b).
Fig. 5.
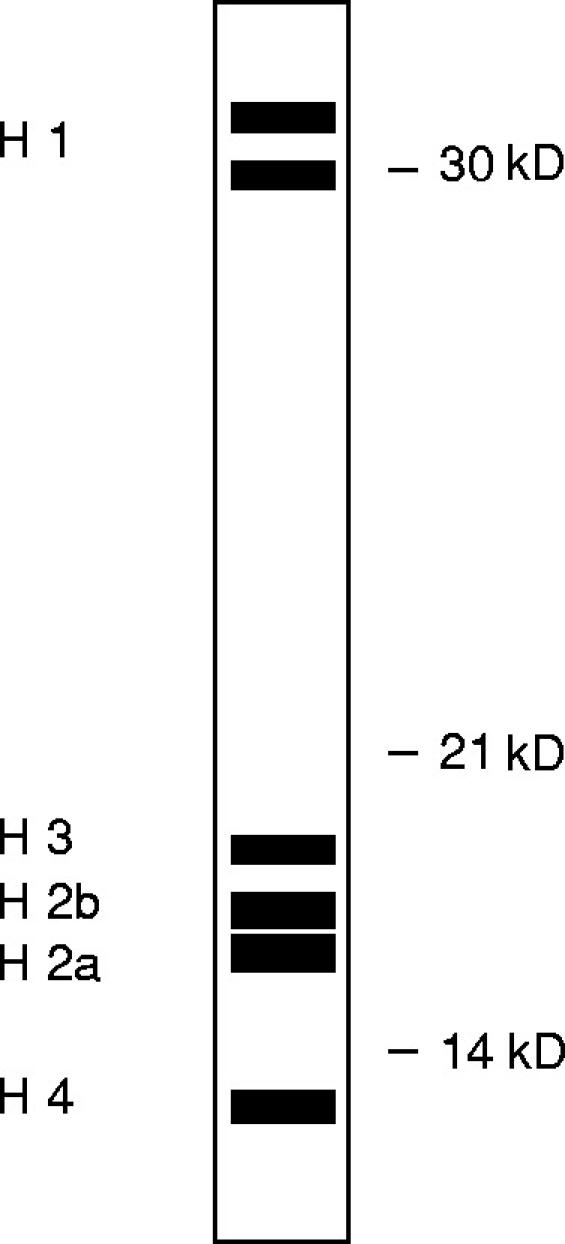
Diagrammatic representation of histone subunits as would be seen on an SDS–PAGE gel.
Fig. 6.
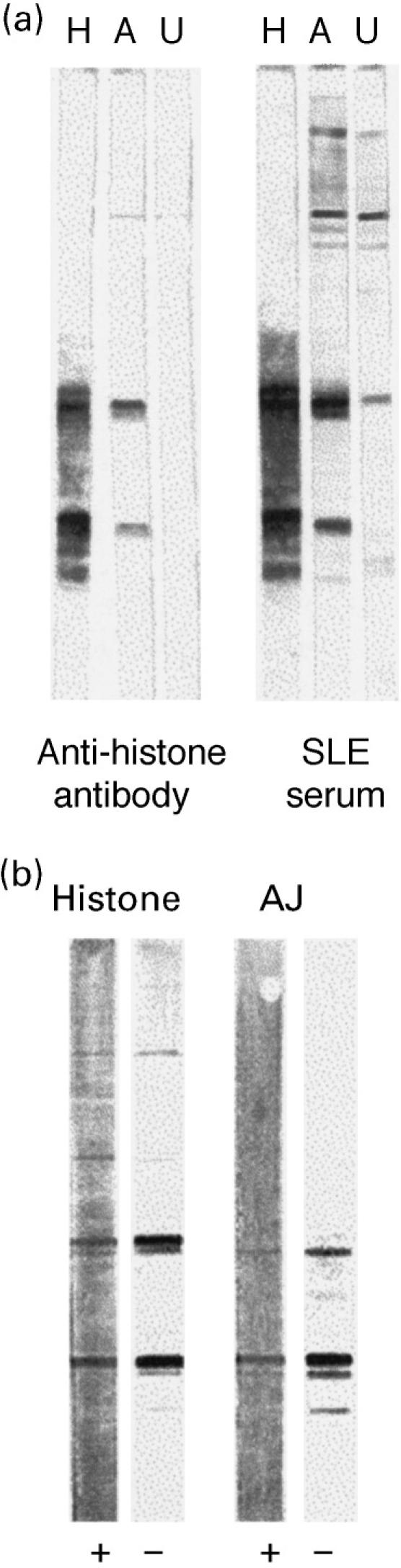
Western blots that compare staining of apoptotic Jurkat cell lysate with histone. (a) Staining of purified histone (H), UV-irradiated Jurkat cells lysate (A) and untreated Jurkat cell lysate (U) with anti-histone antibody and SLE serum. (b) Staining of histone and apoptotic Jurkat (AJ) preparations with anti-histone antibody (−) is reduced by preincubation of antibody with purified histones (+).
The identification of the prominent bands as histones is consistent with their presence in the soluble lysate fraction of the apoptotic Jurkat cells in which the nuclei had been degraded and nucleosomes released, and their absence from the blots of the untreated Jurkat cells from which the nuclear material would be in the insoluble pelleted fraction. Other proteins were altered as expected by the induction of apoptosis: for example, an additional 40-kD band was detected by the commercially obtained human serum containing antibodies to U1-RNP (Fig. 4). This is an apoptosis-related cleavage product of 70-kD U1-RNP, as observed by Casciola-Rosen et al. [4]. It was apparent that this human serum containing anti-U1-RNP antibodies also recognized the histones (Fig. 4).
Release of histones from apoptotic U937 cells
Lysates of U937 cells were prepared in the same manner as for Jurkat cells and Western blotted with some of the SLE sera. As found with Jurkat, no histone bands were detected when probing the soluble lysate fraction of untreated U937 cells (data not shown). Similar IgG antibody staining of the 21-kD and 13-kD histone bands was obtained with both the apoptotic U937 and apoptotic Jurkat soluble lysate fractions produced from UV-irradiated cells (Fig. 7a; position of the histone bands marked by arrows). However, in contrast to the results with Jurkat cells, the 33-kD H1 histone band was extremely weak or undetectable on the Western blots of the U937 soluble lysate fractions (Fig. 7a). This observation was reproducible in separate experiments using different batches of apoptotic Jurkat and U937 cell lysates. Furthermore, this result was not due merely to generally weaker staining of U937 bands since, for example, the 13-kD band was much weaker than the 33-kD band on the Western blots of the Jurkat lysates, but the 13-kD band was clearly detectable on the Western blots of the U937 lysates (Fig. 7a).
Fig. 7.
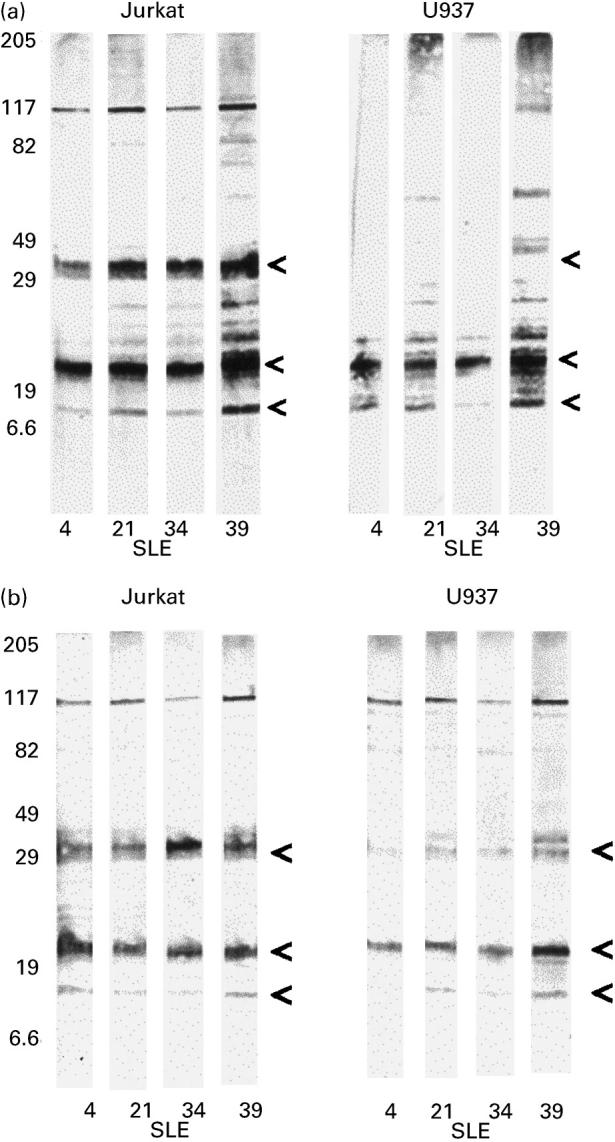
Western blots showing a difference in staining of apoptotic Jurkat cell lysates and U937 cell lysates with SLE sera. The soluble antigens from supernatant of Triton X-100-lysed cells (a) showed the 33-kD band in apoptotic Jurkat preparations, but this band was very weak or absent from the apoptotic U937 cell preparations. The total cell contents were lysed with reducing sample buffer (b), resulting in the previously absent band being weakly detected in the U937 cells. Prominent bands are indicated by arrows.
In order to confirm that histone H1 is a component of U937 cells, although it is barely present in the soluble apoptotic material, equivalent numbers of untreated U937 and Jurkat cells were lysed directly in SDS–PAGE sample buffer. This would solublize all cellular constituents, including the nuclear material, in contrast to the milder lysis conditions used in the previous experiments. Western blotting of these cell lysates probed with SLE IgG serum antibodies showed very similar bands for U937 and Jurkat, including the presence of the 33-kD H1 histone band (Fig. 7b), although this band for U937 tended to be weaker.
DISCUSSION
The results presented here indicate that protein autoantigens can be differentially released by different cell types undergoing apoptosis. Using different sources of apoptotic material we have shown that IgG antibodies in SLE patients' sera can distinguish several proteins revealed during apoptosis. Some of these antigens were also recognized by IgG antibodies in normal sera, though less strongly. The SLE sera were also able to detect antigens in untreated viable cells that were not seen by the normal sera. These observations are consistent with previous reports that constitutive cellular autoantigens become more accessible in cells undergoing apoptosis [2], and that several of the autoantigens are cleaved by caspase enzymes, potentially exposing cryptic epitopes [4,5].
We have demonstrated that apoptotic cells are a source of soluble antigen, not seen in preparations from untreated cells. The proteins that were consistently detected were histone subunits: the pattern of staining by SLE sera was almost identical to that of an anti-histone antibody, and could be abrogated by the absorption of the sera with histones. Histones are major components of the nucleosome unit of chromatin, and cleavage of chromatin to release nucleosomes is a key event during apoptosis. Consistent with this, we found that the histones of non-apoptotic cells were not soluble under mild lysis conditions which would not dissolve chromatin, but were soluble when apoptotic cells were exposed to the same conditions. Antibodies to nucleosomes, DNA and histones are a common feature of SLE, and levels of anti-nucleosome antibodies correlate with levels of circulating nucleosomes [13]. Also, histone cleavage can occur during apoptosis, particularly affecting H3 [12]. Antibodies are produced which are specific for individual histones [7,8] as well as complexes, particularly (H2A-H2B)–DNA [9]. However, we were unlikely to have such complexes in the gel because of the denaturing conditions of the gel electrophoresis, and DNA, or DNA–protein complexes, have a high molecular weight and would not have entered the gel. This also excludes the possibility that anti-DNA antibodies in the SLE sera were cross-reacting with histone–DNA complexes. However, it is possible that some of the serum reactivity with the histones on the Western blots was not due to histone-specific antibodies, but rather resulted from DNA–anti-DNA immune complexes in the sera binding because of the affinity of acidic DNA for basic histones. Such interactions have been reported previously [14]. We cannot totally exclude this as a contributory factor in our experiments, but it is not supported by our finding that treatment of SLE sera (21, 34 and 39) according to a standard protocol to precipitate immune complexes with polyethylene glycol [15] did not remove the reactivity with histones on Western blots (data not shown). In any case, the availability of nucleosomal material from apoptotic cells could stimulate the production of antibodies to both histones and DNA [16]
A variety of assays has been described for the detection of antibodies to histones in SLE, particularly ELISAs, but also immunoblotting as employed in this study (reviewed in [7] and [8]). For clinical utility, assay conditions are normally adjusted to maximize the difference between antibody detection in patients and normal controls. However, our experiments indicate that IgG anti-histone antibodies are present in the sera of many normal individuals, although at lower levels than in SLE patients. In view of the variability of these reactions with sera from different individuals (Fig. 3), it seems unlikely that they are merely due to non-specific charge interactions between the highly basic histones and relatively acidic immunoglobulins, but are indicative of specific antibody–antigen interactions. Furthermore, others have reported that histone-specific antibodies are present in normal individuals [17] and that mitogen stimulation of normal peripheral blood mononuclear cells (PBMC) induces the production of anti-histone antibodies [18]. In addition, histone-specific T cell lines can be generated from normal individuals as well as SLE patients [11]. These findings are consistent with a degree of autoreactivity being neither unusual nor harmful, but that it is the dysregulation of these processes which may lead to autoimmune disease.
SLE patients vary widely in the occurrence and levels of autoantibodies to individual cellular constituents, both between patients and at different times in the same patient. With regard to histone-specific antibodies it has been observed, for example, that different patients can show a predominance of antibodies to either H1 and H2B, or to H2A + H4 [19]. If, as has been hypothesized, material from apoptotic cells drives the autoimmune response, then this variability may relate to the availability and nature of the apoptotic material. This could be affected by several factors, including the stimulus for apoptosis and the cell type in which apoptosis is induced. In our experiments looking particularly at histones we observed little difference in the availability of autoantigens derived from Jurkat cells induced to undergo apoptosis by different stimuli (UV radiation, cyclohexamide or Fas ligation). This is consistent with other reports that different inducers of apoptosis (UV irradiation, anti-Fas antibody, granzyme B) have similar effects on antigens such as PARP, DNA-PK, U1-RNP [20] because these inducers all indirectly trigger the activation of caspase enzymes that cleave these antigens. By contrast, we did see a clear difference in the exposure of autoantigens between the different types of cell line employed, with a 33-kD protein consistent with histone H1 being solubilized during apoptosis of Jurkat cells but not U937 cells (Fig. 7a). This specific observation suggests the more general hypothesis that differences in the main apoptotic cell types acting as sources of autoantigens in different SLE patients may relate to the heterogeneity of autoantibody production and, possibly, of clinical manifestations.
It is at present unclear why histone H1 (but not the other histones) is largely absent from the soluble lysate fraction of apoptotic U937 cells. This finding is in some ways surprising, since H1 is external to the nucleosome core and therefore might be expected to be the most accessible of the histones. Furthermore, complete solubilization of untreated U937 cells in SDS–PAGE sample buffer showed that these cells do possess histone H1 which reacts with IgG antibodies in SLE sera (Fig. 7b). This 33-kD band is however generally weaker for U937 cells than for Jurkat cells, which may be indicative of a fundamental difference between these cell lines. One possibility is suggested by the report of H1 binding to calreticulin [21]: thus, H1 may bind to other cellular components in apoptotic U937 cells (but not apoptotic Jurkat cells) that remain insoluble under mild lysis conditions. Alternatively, H1 may undergo degradation during apoptosis of U937 cells, but remain intact in apoptotic Jurkat cells.
Apoptosis is a normal physiological process involved in the control of cell populations and the removal of unwanted or exhausted cells. However, abnormalities in the way the body deals with apoptosis may be relevant to the development of SLE. Thus, inefficient removal of apoptotic debris may result in damage to surrounding tissue by harmful cell contents leading to inflammation, and may also result in increased exposure of potential immunogens: both could perpetuate an autoimmune response (reviewed in [22]). Macrophages normally play an important role in the non-immunogenic clearance of apoptotic cells, but it has been reported that macrophages of SLE patients show impaired phagocytosis of apoptotic cells [23]. This could make apoptotic material available both for binding by autoreactive B cells specific for cellular constituents such as nucleosomes, and for phagocytosis by dendritic cells. The apoptotic material could then be immunogenically presented to T cells [24]. This may happen in a limited and controlled way in normal individuals, which would explain the low levels of histone-specific antibodies in normal subjects which we have observed. However, it may happen in an exaggerated way in SLE and/or occur in an environment of autoreactive T cell dysregulation.
Environmental triggers such as UV light can induce a systemic flare in photosensitive SLE patients. We have used UV light as an inducer of apoptosis and demonstrated that soluble antigens are revealed during apoptosis. Apoptosis of keratinocytes by UV light induces skin lesions and the antigens that form in the blebs of these apoptotic cells can stimulate autoantibody production [25]. Furthermore, increased apoptosis, as a result of inflammation during a lupus flare, may enhance anti-histone antibody production due to the increased availability of antigen, thereby perpetuating the disease.
In conclusion, different triggers of apoptosis promote the availability of similar autoantigens. Histones are a major component of these antigens, and are strongly recognized by autoantibodies in SLE sera. Different apoptotic cell types can vary in the types of autoantigens released, which may be relevant to the variety of autoantibodies and clinical manifestations in SLE.
Acknowledgments
This work was funded by the Jones Charitable Trust.
REFERENCES
- 1.Stollar B. The antigenic potential and specificity of nucleic acids, nucleoproteins, and their modified derivatives. Arthritis Rheum. 1981;28:1010–7. doi: 10.1002/art.1780240806. [DOI] [PubMed] [Google Scholar]
- 2.Casciola-Rosen L, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–30. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casciola-Rosen L, Miller D, Anhalt G, et al. Specific cleavage of the 70kDa protein component of the U1-small nuclear ribonucleoprotein is a characteristic biochemical feature of apoptotic cell death. J Biol Chem. 1994;269:30757–60. [PubMed] [Google Scholar]
- 4.Casciola-Rosen L, Anhalt G, Rosen A. DNA-dependent protein kinase is one of a subset of autoantigens specifically cleaved early during apoptosis. J Exp Med. 1995;182:1625–34. doi: 10.1084/jem.182.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casiano C, Martin S, Green D, et al. Selective cleavage of nuclear autoantigens during CD95 (Fas/APO-1)-mediated T cell apoptosis. J Exp Med. 1996;184:765–70. doi: 10.1084/jem.184.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enari M, Sakahira H, Yokoyama H, et al. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 7.Stemmer C, Muller S. Histone autoantibodies other than (H2A-H2B)-DNA autoantibodies. In: Peter J, Shoenfeld Y, editors. Autoantibodies. Amsterdam: Elsevier Science B. V.; 1996. pp. 373–84. [Google Scholar]
- 8.Fritzler M. Antibodies to histone and the role of the nucleosome. In: Wallace DJ, Hahn BH, editors. Dubois' lupus erythematosus. Baltimore: Williams & Wilkins; 1997. pp. 423–41. [Google Scholar]
- 9.Rubin R. Histone (H2A-H2B)-DNA autoantibodies. In: Peter J, Shoenfeld Y, editors. Autoantibodies. Amsterdam: Elsevier Science BV; 1996. pp. 364–71. [Google Scholar]
- 10.Desai-Mehta A, Mao C, Rajagopalan S, et al. Structure and specificity of T cell receptors expressed by potentially pathogenic anti-DNA autoantibody-inducing T cells in human lupus. J Clin Invest. 1995;95:531–41. doi: 10.1172/JCI117695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voll R, Roth E, Girkontaite I, et al. Histone-specific Th0 and Th1 clones derived from systemic lupus erythematosus patients induce double-stranded DNA antibody production. Arthritis Rheum. 1997;40:2162–71. doi: 10.1002/art.1780401210. [DOI] [PubMed] [Google Scholar]
- 12.Cabrespines A, Laderach D, Lebosse C, et al. Isolation and characterization of apoptotic nucleosomes, free and complexed with lupus autoantibody generated during hybridoma B cell apoptosis. J Autoimmun. 1998;11:19–27. doi: 10.1006/jaut.1997.0172. [DOI] [PubMed] [Google Scholar]
- 13.Amoura Z, Piette J, Chabre H, et al. Circulating plasma levels of nucleosomes in patients with systemic lupus erythematosus—correlation with serum antinucleosome antibody titers and absence of clear association with disease activity. Arthritis Rheum. 1997;40:2217–25. doi: 10.1002/art.1780401217. [DOI] [PubMed] [Google Scholar]
- 14.Subiza JL, Caturla A, Pascual-Salcedo D, et al. DNA–anti-DNA complexes account for part of the anti-histone activity found in patients with systemic lupus erythematosus. Arthritis Rheum. 1989;32:406–11. doi: 10.1002/anr.1780320409. [DOI] [PubMed] [Google Scholar]
- 15.Zubler PH, Perrin LH, Greighton WD. Use of polyethylene glycol (PEG) to concentrate immune complexes from serum or plasma samples. Ann Rheum Dis. 1977;36:23–25. [Google Scholar]
- 16.Amoura Z, Piette J-C, Bach J-F, et al. The key role of nucleosomes in lupus. Arthritis Rheum. 1999;42:833–43. doi: 10.1002/1529-0131(199905)42:5<833::AID-ANR1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 17.Bustos A, Boimorto R, Subiza J, et al. Inhibition of histone/anti-histone reactivity by histone binding serum components; differential effect on anti-H1 versus anti-H2B antibodies. Clin Exp Immunol. 1994;95:408–14. doi: 10.1111/j.1365-2249.1994.tb07011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Dell JR, Bizar-Schneebaum A, Kotzin BL. In vitro anti-histone antibody production by peripheral blood cells from patients with systemic lupus erythematosus. Clin Immunol Immunopathol. 1988;47:343–53. doi: 10.1016/s0090-1229(88)80011-4. [DOI] [PubMed] [Google Scholar]
- 19.Kohda S, Kanayam Y, Okamura M, et al. Clinical significance of antibodies to histones in systemic lupus erythematosus. J Rheumatol. 1989;16:24–28. [PubMed] [Google Scholar]
- 20.Andrade F, Roy S, Nicholson D, et al. Granzyme-B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity. 1998;8:451–60. doi: 10.1016/s1074-7613(00)80550-6. [DOI] [PubMed] [Google Scholar]
- 21.Treves S, Bajocchi G, Zorzato F, et al. Identification and characterization of a calreticulin-binding nuclear protein as histone (H1), an autoantigen in systemic lupus erythematosus. Lupus. 1998;7:479–87. doi: 10.1191/096120398678920505. [DOI] [PubMed] [Google Scholar]
- 22.Ren Y, Savill J. Apoptosis: the importance of being eaten. Cell Death Diff. 1998;5:563–8. doi: 10.1038/sj.cdd.4400407. [DOI] [PubMed] [Google Scholar]
- 23.Herrmann M, Voll R, Zoller D, et al. Impaired phagocytosis apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–50. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 24.Albert M, Pearce S, Francisco L, et al. Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. 1998;7:1359–68. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casciola-Rosen L, Rosen A. Ultraviolet light-induced keratinocyte apoptosis: a potential mechanism for the induction of skin lesions and autoantibody production in LE. Lupus. 1997;6:175–80. doi: 10.1177/096120339700600213. [DOI] [PubMed] [Google Scholar]