

Abstract
This study was designed to investigate VEGF production from peripheral blood mononuclear cells (PBMC) from patients with rheumatoid arthritis (RA) compared with healthy controls and to identify the predominant cellular source in PBMC isolated from RA patients. The regulation of PBMC VEGF production by cytokines and synovial fluid (SF) was studied. PBMC were isolated from RA patients and healthy controls and stimulated with lipopolysaccharide (LPS), IL-1β, IL-4, IL-6, IL-8, IL-10, TNF-α and transforming growth factor-beta (TGF-β) isoforms for varying time points up to 72 h at 37°C/5% CO2. The effect of SF on VEGF secretion by PBMC was also studied. Supernatant VEGF levels were measured using a flt-1 receptor capture ELISA. RA patients had significantly higher spontaneous production of VEGF compared with controls, and monocytes were identified as the predominant cellular source. RA PBMC VEGF production was up-regulated by TGF-β isoforms and TNF-α and down-regulated by IL-4 and IL-10, with no effect observed with IL-1β, IL-6 and IL-8. Antibody blocking experiments confirmed that TNF-α and not TGF-β isoforms in SF increased VEGF secretion by RA PBMC. These results emphasize the importance of monocytes as a source of VEGF in the pathophysiology of RA. Several cytokines known to be present in SF can modulate the level of VEGF secretion, but the predominant effect of SF in VEGF up-regulation is shown to be dependent on TNF-α.
Keywords: VEGF, monocytes, cytokines, rheumatoid arthritis
INTRODUCTION
The macrophage has been proposed to play an important role in the pathogenesis of disease progression in rheumatoid arthritis (RA). Histological examination of affected synovium reveals evidence of macrophage infiltration, with a significant correlation existing between the degree of infiltration and disease activity [1, 2]. Macrophages in synovial tissue are in an activated state, with increased expression of MHC class II molecules [3] and the β-integrins CR3 (CD11b/CD18) and CR4 (CD11c/CD18) [4], and are known to produce a range of cytokines including IL-1β [5] and TNF-α [6].
VEGF is a disulphide-linked homodimer of 34–42 kD. It is an endothelial cell mitogen [7], which promotes angiogenesis and also has potent vascular permeability-enhancing properties [8]. Alternate splicing of mRNA results in the generation of four protein species of 121, 165, 189 and 206 amino acids. On the basis of protein structure, particularly the pattern of conserved cysteines and sequence homology, VEGF is considered to be a member of the platelet-derived growth factor (PDGF) protein family [9], which also includes the more recently described placental growth factor (PlGF), VEGF-B, and VEGF-C [10–12]. VEGF mediates biological function by interaction with two specific tyrosine kinase receptors; flt-1 [13] and KDR [14] (also known as VEGFR1 and VEGFR2, respectively). These receptors are expressed predominantly on endothelial cells in keeping with the endothelial cell-specific roles for VEGF, although circulating monocytes and neutrophils have also been shown to express flt-1, which may mediate VEGF-induced cell chemotaxis [15]. A number of cytokines and growth factors are known to up-regulate VEGF expression, including IL-1β [16], PDGF-β [17], transforming growth factor-beta (TGF-β) [18, 19], basic fibroblast growth factor (bFGF) [20], epidermal growth factor (EGF) [21], TGF-α [22] and angiotensin II [23].
There have been several publications reporting dysregulated VEGF expression in RA, suggesting a potential role for VEGF in the disease pathogenesis. High levels of VEGF have been detected in synovial fluid (SF) by ELISA, macrophages lining the synovium have been shown to express VEGF mRNA strongly, and the microvascular endothelial cells of nearby blood vessels also express mRNA for flt-1 and KDR [24–26]. VEGF polypeptide expression has been demonstrated in subsynovial macrophages, fibroblasts surrounding microvessels, vascular smooth muscle cells and synovial lining cells [27]. It has been hypothesized that synovial macrophages are stimulated to synthesize and secrete VEGF, which binds to receptors on local endothelium, initiating angiogenesis and migration into the joint cavity, with a resultant increase in vascular permeability and leakage of plasma proteins into the joint space [24].
The aims of this study were (i) to determine the levels of spontaneous and stimulated VEGF production by peripheral blood mononuclear cells (PBMC) from patients with RA and healthy control subjects, (ii) to investigate the regulation of monocyte VEGF production by cytokines proposed as having a role in the initiation and progression of RA, and (iii) to determine the effects of SF on VEGF production by PBMC.
PATIENTS AND METHODS
Materials
Recombinant VEGF165 and sflt-1 were gifts from Zeneca Pharmaceuticals (Alderley Edge, UK). Peroxidase-conjugated anti-rabbit antibody was purchased from Jackson ImmunoResearch (Luton, UK). Recombinant human IL-1β, IL-4, IL-6, IL-8, IL-10, TGF-β 1 and TGF-β 3 and neutralizing antibodies to IL-4 and IL-10 were purchased from R&D Systems (Abingdon, UK). Recombinant human TNF-α and TGF-β 2 and blocking antibody to TGF-β 1,2,3 were purchased from Genzyme Diagnostics (West Malling, UK). Blocking antibody to TNF-α was from Serotec (Oxford, UK). Lipopolysaccharide (LPS; Escherichia coli-derived) was purchased from Quadratech (Epsom, UK). Lymphocyte separation medium (Lymphoprep) was from Flow Labs (Irvine, UK).
Patients and samples
Blood samples were collected from patients with RA during routine follow up and also from healthy adult volunteers. RA patients fulfilled the ACR diagnostic criteria. Whole blood was collected into EDTA blood tubes. The total and differential leucocyte counts on the patient samples were within the normal range. SF were obtained by joint aspiration performed as part of routine clinical management from patients with a variety of arthropathies; fluids were diagnosed according to SF pathology alone [28] and categorized into RA, osteoarthritis (OA), primary inflammatory (PI) arthropathies or non-inflammatory (NI) arthropathies. Approval for the study was granted by the Central Manchester Ethics Committee.
Secretion of VEGF by PBMC isolated from RA patients and normal controls
PBMC from RA patients (n = 21, 18 female and three male, age 51 ± 8 years (mean ± s.d.)) and healthy controls (n = 21, 11 female and 10 male, age 35 ± 7 years) were isolated from blood [29], resuspended at 2 × 106/ml in RPMI 1640/10% fetal calf serum (FCS) and incubated for 24 h and 72 h in the presence of LPS (10 U/ml), with unstimulated cells acting as controls.
VEGF levels in cell culture supernatant samples were measured using an in-house sflt-1 receptor capture ELISA previously described in detail [30]. This assay has been shown to measure free VEGF and not VEGF complexed to flt-1. Intra-and interplate coefficients of variation for the assay were 4.5% and 11.5%, respectively.
Secretion of VEGF by PBMC, monocytes and lymphocytes isolated from RA patients
Lymphocytes and monocytes were isolated from the PBMC samples of five RA patients by monocyte depletion using an anti-CD14 antibody-coated magnetic bead (miniMACS, Bisley, UK) system. The separate populations of monocyte-depleted lymphocytes and purified monocytes were analysed by flow cytometry (Coulter Epics XL, Luton, UK) using size and granularity to confirm purity. Cells were resuspended at 2 × 106/ml in RPMI 1640/10% FCS and cultured for 24 h and 72 h at 37°C/5% CO2 in the presence of LPS (10 U/ml) with unstimulated cells acting as controls. VEGF levels were quantified in the culture supernatants by ELISA.
Cytokine regulation of VEGF secretion by PBMC isolated from RA patients
Experiments were performed to investigate a potential role for cytokines in the control of spontaneous PBMC VEGF secretion. PBMC (2 × 106/ml) isolated from 10 RA patients were incubated with a range of cytokines including IL-1β, IL-4, IL-6, IL-8, IL-10, TNF-α, TGF-β 1, TGF-β 2 and TGF-β 3 at concentrations of 0.01, 0.1, 1, 10 and 100 ng/ml for 24 h. Supernatants were then assayed for VEGF by ELISA. Time course experiments were also performed on PBMC isolated from three RA patients by adding TGF-β 1 (1 ng/ml) and quantifying VEGF levels at 1, 2, 4, 8 and 24 h after its addition. Results were expressed as a percentage of the level of VEGF in unstimulated control cultures run in conjunction with each experiment.
Further experiments were carried out to determine whether TNF-α-induced IL-4 or IL-10 secretion by PBMC influenced VEGF secretion. PBMC isolated from eight RA patients were cultured with 10 ng/ml rTNF-α and 1 μg/ml of neutralizing antibody to IL-4 (R&D systems) or IL-10 (R&D systems) for 24 h. Cells stimulated with 10 ng/ml rTNF-α alone under the same conditions acted as control.
Effect of SF on VEGF secretion by PBMC
To investigate the potential of SF to modulate VEGF production, 1 × 106 PBMC isolated from normal adults resuspended in 450 μl of RPMI 1640 were incubated with 50 μl of SF (10%) from 19 RA patients, 12 PI patients, six OA patients and 10 NI patients for 24 h at 37°C/5% CO2. Cells in RPMI 1640/10% FCS acted as control. Blocking studies were performed using MoAbs to TNF-α and TGF-β 1,2,3. PBMC from a normal healthy volunteer (1 × 106) were incubated with 10% SF from 10 RA patients in RPMI 1640 with no FCS, alone or with 20 μg/ml of anti-TNF-α, anti-TGF-β 1,2,3 or mouse immunoglobulin as control in a total volume of 500 μl for 24 h at 37°C/5% CO2. Cells only and cells with each antibody were also included as controls.
RESULTS
Secretion of VEGF by PBMC isolated from RA patients and controls
PBMC from RA patients spontaneously secreted significantly more VEGF compared with cells from control subjects at 24 h and 72 h (P < 0.02, unpaired t-test) (Fig. 1). Stimulated RA PBMC also secreted significantly more VEGF than stimulated control cells at 72 h (P = 0.008, unpaired t-test) with no significant difference at 24 h. All VEGF levels at 72 h were significantly higher than the corresponding 24 h levels. Patient samples and control samples were not age- or sex-matched, but unstimulated VEGF production at 24 h in 45 RA patients was found not to correlate with age (P = 0.9) or be affected by gender (Pearson correlation).
Fig. 1.

Levels of VEGF secretion by unstimulated and stimulated (10 U/ml lipopolysaccharide (LPS)) peripheral blood mononuclear cells (PBMC) isolated from 21 rheumatoid arthritis (RA) patients (▪) and 21 healthy controls (□) after 24 h and 72 h in culture. Spontaneous secretion of VEGF by RA patients was significantly higher after 24 h and 72 h compared with controls. *P < 0.02 (unpaired t-test).
Secretion of VEGF protein by PBMC, monocytes and lymphocytes isolated from RA patients
After 24 h in culture, CD14 magnetic bead-purified unstimulated monocytes isolated from five RA patients secreted similar levels of VEGF compared with the PBMC population from which they were isolated, with the lymphocytes secreting a much lower level (Table 1). The 24 h LPS-stimulated corresponding populations exhibited a similar pattern of secretion. After 72 h, results again followed a similar pattern as those at 24 h. These results demonstrate the monocyte as the main source of VEGF in PBMC cultures. Monocyte and lymphocyte populations were all shown to have > 95% purity as determined by flow cytometry.
Table 1.
Levels of VEGF secretion by unstimulated (resting) and stimulated (10 U/ml lipopolysaccharide (LPS)) peripheral blood mononuclear cells (PBMC), lymphocytes and CD14 magnetic bead-purified monocytes isolated from five rheumatoid arthritis (RA) patients after 24 h and 72 h in culture. VEGF secretion by monocytes was not significantly different form VEGF secretion by PBMC at all time points.

Cytokine regulation of VEGF secretion by PBMC isolated from RA patients
TGF-β 1 up-regulated VEGF production from PBMC isolated from 10 patients with RA in a dose-dependent manner, with maximal production at 1 ng/ml (P < 0.05, one-way anova with Bonferroni correction) (Fig. 2). Similar results were also observed with TGF-β 2 and TGF-β 3 (data not shown). Time-course experiments revealed that VEGF production in response to TGF-β 1 in this system was maximal between 8 h and 24 h (data not shown). TNF-α also up-regulated VEGF production in a dose-dependent manner, with maximal production at 10 ng/ml (P < 0.05, one-way anova with Bonferroni correction) (Fig. 2).
Fig. 2.

Levels of VEGF secretion from peripheral blood mononuclear cells (PBMC) isolated from 10 rheumatoid arthritis (RA) patients in response to transforming growth factor-beta 1 (TGF-β1) (a) and TNF-α (b) at 100, 10, 1, 0.1 and 0.01 ng/ml after 24 h in culture. *P < 0.05 compared with control (one-way anova with Bonferroni correction).
Both IL-4 and IL-10 down-regulated VEGF production from PBMC isolated from patients with RA compared with unstimulated cells in a dose-dependent manner, with 1, 10 and 100 ng/ml of IL-10 and 10 and 100 ng/ml of IL-4 having a significant effect (P < 0.05, one-way anova with Bonferroni correction) (Fig. 3). Addition of IL-4 and IL-10 together had no synergistic effect on the inhibition of VEGF secretion compared with their addition alone at the same concentrations (data not shown). IL-1β, IL-6, and IL-8 had no significant effect on RA PBMC VEGF production.
Fig. 3.
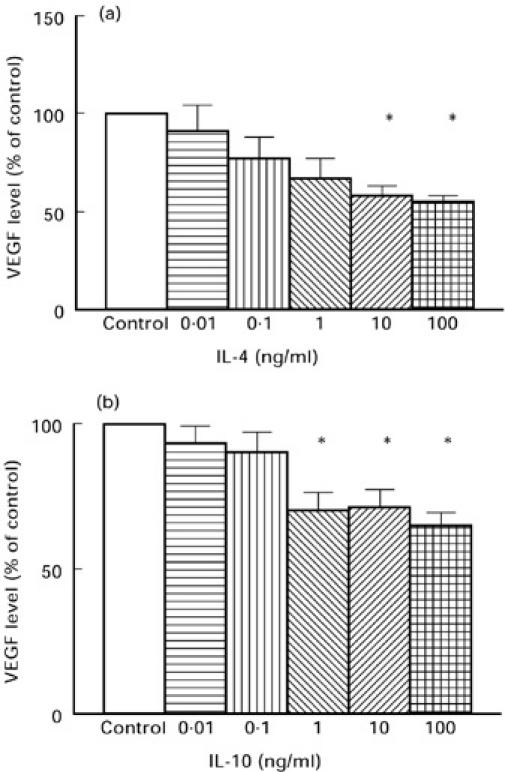
Levels of VEGF secretion from peripheral blood mononuclear cells (PBMC) isolated from 10 rheumatoid arthritis (RA) patients in response to IL-4 (a) and IL-10 (b) at 100, 10, 1, 0.1 and 0.01 ng/ml after 24 h in culture. *P < 0.05 compared with control (one-way anova with Bonferroni correction).
Inhibition of IL-4 or IL-10 activity with neutralizing MoAbs had no statistically significant effect on TNF-α-induced expression of VEGF (mean ± s.e.m. VEGF secretion: 10 ng/nl TNF-α alone 786 ± 256 pg/ml, 10 ng/nl TNF-α with anti-IL-4 791 ± 204 pg/ml, and 10 ng/nl TNF-α with anti-IL-10 797 ± 221 pg/ml).
Effect of SF on VEGF secretion by PBMC
The incubation of SF from patients with a variety of arthropathies with PBMC, overall resulted in enhanced VEGF production in all disease groups (Fig. 4a). RA SF resulted in the largest increase in VEGF secretion compared with cells with 10% FCS alone, but there was no significant difference between the disease groups.
Figure 4.
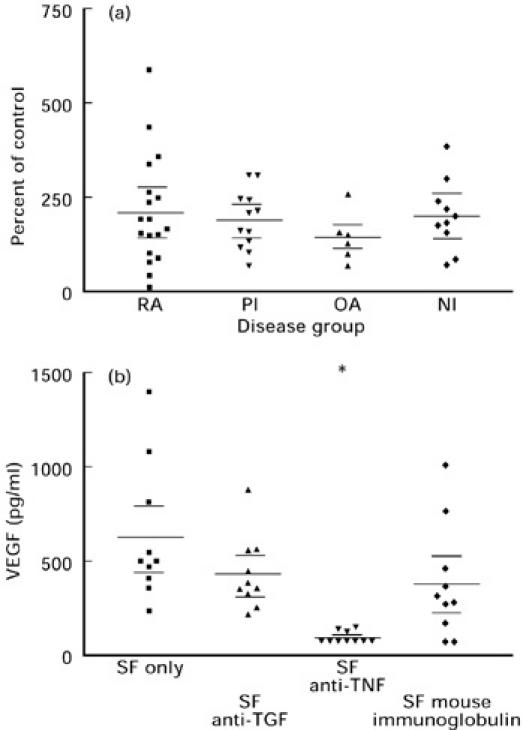
The effect of 10% synovial fluid (SF) from rheumatoid arthritis (RA), primary inflammatory (PI), osteoarthritis (OA) and non-inflammatory (NI) groups on VEGF secretion by normal peripheral blood mononuclear cells (PBMC) (a). The results are expressed as a percentage of VEGF secretion by cells alone and the VEGF already present in the SF was corrected for by subtracting 10% of the neat SF VEGF level. No significant differences were observed between groups. The effect of 10% RA SF (n = 10) alone, or in combination with 20 μg/ml of anti-TNF-α, anti-transforming growth factor-beta 1,2,3 (TGF-β1,2,3) or mouse immunoglobulin as control on VEGF secretion by normal PBMC after 24 h in culture (b). *P < 0.001 compared with cells with SF alone (one-way anova with Bonferroni correction).
Neutralizing TGF-β isoforms in SF resulted in a mean reduction of 30% in VEGF secretion compared with cells incubated with SF only (means 436 pg/ml and 627 pg/ml, respectively) (Fig. 4b). This was comparable to the levels of VEGF secretion obtained with mouse immunoglobulin as an antibody control (39% reduction). Inhibition of TNF-α, however, resulted in significant inhibition of VEGF secretion (P < 0.001, one-way anova with Bonferroni correction) by > 84% with seven out of the 10 samples having undetectable levels of VEGF at the end of 24 h.
DISCUSSION
In this study, unstimulated PBMC isolated from RA patients secreted significantly more VEGF after 24 h and 72 h in culture compared with cells from healthy controls. LPS stimulation produced less up-regulation of RA PBMC VEGF production than that observed with control PBMC, which could be due to RA PBMC being in a higher activation state than normal PBMC. This constitutively higher expression of VEGF by RA PBMC may be a feature of inflammatory joint disease, as similar experiments on PBMC isolated from patients with glomerulopathies revealed no significant differences between spontaneous VEGF secretion compared with control subjects (P. Brenchley, unpublished observation).
The experimental data presented show the monocyte to be the source of VEGF from PBMC stimulated by LPS. Interestingly, T lymphocytes have been shown to express VEGF mRNA [31] and to secrete protein [32]. Monocytes, and especially macrophages, have previously been shown to express VEGF protein by immunohistochemistry in RA synovial tissue [24–27] and placental macrophages (Hoffbauer cells) [33].
TGF-β isoforms and TNF-α were the only cytokines which up-regulated VEGF protein expression by RA PBMC within 24 h. Previous publications have reported that TGF-β can up-regulate VEGF expression in glioma cells, vascular smooth muscle cells and keratinocytes [34, 35] and down-regulate expression of flk-1, the rat equivalent of the VEGF receptor KDR [36]. Time-course experiments revealed that increased VEGF production occurred between 8 h and 24 h of TGF-β 1 stimulation, suggesting that TGF-β 1 acts by stimulating transcription of new VEGF mRNA. Like TGF-β, TNF-α is thought to be a cytokine with angiogenic properties [37]. Our demonstration of the direct up-regulation of VEGF by TNF-α may explain this function. TNF-α has previously been shown to induce VEGF mRNA expression in human glioma cells through the transcription factor SP-1, although VEGF protein levels were not measured [38]. The beneficial effects of blocking TNF-α by antibody therapy in RA [39, 40] may therefore be partly explained by the potential of TNF-α to up-regulate VEGF expression in monocytes found within the SF and synovial tissue. Anti-TNF-α therapy may be blocking an important feedback loop, since monocytes and macrophages are the main source of TNF-α and VEGF within the RA joint [41].
IL-4 inhibits the LPS-induced IL-1, TNF-α, prostaglandin E2(PGE2) and 92-kD gelatinase production in human monocytes [42, 43]. IL-10 has also been shown to inhibit IL-1 and TNF-α production by synovial cell cultures [44]. In this study, IL-4 and IL-10 at physiological concentrations were observed to down-regulate VEGF secretion by PBMC and monocytes, with IL-4 showing the greater effect. Unlike IL-4, IL-10 has been detected in the peripheral blood [45] and synovial joints of RA patients by reverse transcriptase-polymerase chain reaction (RT-PCR [44], but its action may be counteracted by TGF-β and TNF-α.
The mean effect of SF from four patient groups, RA, OA, PI and NI, was to increase VEGF expression by PBMC, with no significant difference between groups. Blocking studies determined that TNF-α in SF was responsible for the SF induction of VEGF, with TGF-β in SF having little or no effect. While both of these cytokines are abundant in SF [46, 47], these results support the theory that SF TNF-α has the greatest potential to induce angiogenesis within the joint through promotion of VEGF expression. One of the major benefits of TNF-α blockade in RA [39, 40] may be the inhibition of TNF-α-induced VEGF secretion by monocytes.
Acknowledgments
We would like to thank Angela Thompson for her help in sample collection and Dr Don Ogilvie for the recombinant VEGF165 and sflt-1. M.J.B. was funded by the Arthritis Research Council, UK, Project number B0558.
REFERENCES
- 1.Harris ED. Rheumatoid arthritis. Pathophysiology and implications for therapy. N Eng J Med. 1990;322:1277–88. doi: 10.1056/NEJM199005033221805. [DOI] [PubMed] [Google Scholar]
- 2.Yanni G, Whelan A, Feighery C, Bresnihan B. Synovial tissue macrophages and joint erosion in rheumatoid arthritis. Ann Rheum Dis. 1994;54:39–44. doi: 10.1136/ard.53.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janossy G, Panayi G, Duke O, et al. Rheumatoid arthritis: a disease of T lymphocyte macrophage immunoregulation. Lancet. 1981;2:834–42. doi: 10.1016/s0140-6736(81)91107-7. [DOI] [PubMed] [Google Scholar]
- 4.Allen CA, Highton J, Palmer DG. Increased expression of p150.95 and CR3 leukocyte adhesion molecules by mononuclear phagocytes in rheumatoid synovial membranes. Arthritis Rheum. 1989;32:947–54. doi: 10.1002/anr.1780320803. [DOI] [PubMed] [Google Scholar]
- 5.Kirkham BW, Navaro FJ, Corkill MM, et al. Immunohistochemical localisation of interleukin 1 in rheumatoid and osteoarthritis synovial membrane. J Rheumatol. 1989;28(Suppl.2):47. [Google Scholar]
- 6.Chu CQ, Field M, Feldman M, Maini RN. Localisation of tumour necrosis factor α in synovial tissues and at the cartilage–pannus junction in patients with rheumatoid arthritis. Arthritis Rheum. 1991;34:1125–32. doi: 10.1002/art.1780340908. [DOI] [PubMed] [Google Scholar]
- 7.Gospodarowicz D, Abraham JA, Schilling J. Isolation and characterisation of a vascular endothelial cell mitogen produced by pituitary-derived folliculo stellate cells. Proc Natl Acad Sci USA. 1989;86:7311–5. doi: 10.1073/pnas.86.19.7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Senger DR, Galli SS, Dvorak AM, et al. Tumour cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–5. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 9.Conn G, Bayne ML, Soderman DD, et al. Amino acid and cDNA sequences of a vascular endothelial cell mitogen that is homologous to platelet-derived growth factor. Proc Natl Acad Sci USA. 1990;87:2628–32. doi: 10.1073/pnas.87.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olofsson B, Pajusola K, Kaipainen A, et al. Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc Natl Acad Sci USA. 1996;93:2576–81. doi: 10.1073/pnas.93.6.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joukov V, Pajusola K, Kapainen A, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:290–8. [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J, Gray A, Yuan J, et al. Vascular endothelial growth factor-related protein: a ligand and specific activator of the tyrosine kinase receptor Flt4. Proc Natl Acad Sci USA. 1996;93:1988–92. doi: 10.1073/pnas.93.5.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vries C, Escobedo JA, Ueno H, et al. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–91. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 14.Terman BI, Dougher-Vermazen M, Carrion ME, et al. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun. 1992;187:1579–86. doi: 10.1016/0006-291x(92)90483-2. [DOI] [PubMed] [Google Scholar]
- 15.Shen H, Clauus M, Ryan J, et al. Characterization of vascular permeability factor/vascular endothelial growth factor receptors on mononuclear phagocytes. Blood. 1993;81:2767–73. [PubMed] [Google Scholar]
- 16.Li J, Perrella MA, Tsai JC, et al. Induction of vascular endothelial growth factor gene expression by interleukin-1 beta in rat aortic smooth muscle cells. J Biol Chem. 1995;270:308–12. doi: 10.1074/jbc.270.1.308. [DOI] [PubMed] [Google Scholar]
- 17.Brogi E, Wu TG, Namiki A, Isner JM. Indirect angiogenic cytokines upregulate VEGF and bFGF gene expression in vascular smooth muscle cells, whereas hypoxia upregulates VEGF expression only. Circulation. 1990;90:649–52. doi: 10.1161/01.cir.90.2.649. [DOI] [PubMed] [Google Scholar]
- 18.Pertovaara L, Kaipainen A, Mustonen T, et al. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J Biol Chem. 1994;269:6271–4. [PubMed] [Google Scholar]
- 19.Pepper MS, Vassalli JD, Orci L, Montesano R. Biphasic effect of transforming growth factor-beta 1 on in vitro angiogenesis. Exp Cell Res. 1993;204:356–63. doi: 10.1006/excr.1993.1043. [DOI] [PubMed] [Google Scholar]
- 20.Stavri GT, Zachary IC, Baskerville PA, et al. Basic fibroblastic growth factor upregulates the expression of vascular endothelial growth factor in vascular smooth muscle cells: synergistic interaction with hypoxia. Circulation. 1995;92:11–14. doi: 10.1161/01.cir.92.1.11. [DOI] [PubMed] [Google Scholar]
- 21.Goldman CK, Kim J, Wong WL, et al. Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells: a model of glioblastoma multiforme pathophysiology. Mol Biol Cell. 1993;4:121–33. doi: 10.1091/mbc.4.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Detmar M, Yeo KT, Nagy JA, et al. Keratinocyte-derived vascular permeability factor (vascular endothelial growth factor) is a potent mitogen for dermal microvascular endothelial cells. J Invest Dermatol. 1995;105:44–50. doi: 10.1111/1523-1747.ep12312542. [DOI] [PubMed] [Google Scholar]
- 23.Williams B, Baker AQ, Gallacher B, Lodwick D. Angiotensin II increases vascular permeability factor gene expression by human vascular smooth muscle cells. Hypertension. 1995;25:913–7. doi: 10.1161/01.hyp.25.5.913. [DOI] [PubMed] [Google Scholar]
- 24.Fava RA, Olsen NJ, Spencer-Green G, et al. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med. 1994;180:341–6. doi: 10.1084/jem.180.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bottomley MJ, Holt PLJ, Watson CJ, et al. Vascular endothelial growth factor mediates synovial inflammation. Arthritis Rheum. 1996;39:9, S131. [Google Scholar]
- 26.Koch AE, Harlow LA, Haines GK, et al. Vascular endothelial growth factor—a cytokine modulating endothelial function in rheumatoid arthritis. J Immunol. 1994;152:4149–56. [PubMed] [Google Scholar]
- 27.Nagashima M, Yoshino S, Ishiwata T, Asano G. The role of vascular endothelial growth factor in angiogenesis of rheumatoid arthritis. J Rheumatol. 1995;22:1624–30. [PubMed] [Google Scholar]
- 28.Freemont AJ, Denton J. Atlas of synovial fluid cytopathology. Vol. 18. Higham, USA: Kluwer; 1991. [Google Scholar]
- 29.Boyum A. Separation of white blood cells. Nature. 1964;204:793–4. doi: 10.1038/204793a0. [DOI] [PubMed] [Google Scholar]
- 30.Webb NJA, Bottomley MJ, Watson CJ, Brenchley PEC. Vascular endothelial growth factor (VEGF) is released from platelets during blood clotting: implications for measurement of circulating VEGF levels in clinical science. Clin Sci. 1998;94:395–405. doi: 10.1042/cs0940395. [DOI] [PubMed] [Google Scholar]
- 31.Iijima K, Yoshikawa N, Nakamura H. Activation-induced expression of vascular permeability factor by human peripheral T cells: a non-radioisotope semiquantitative reverse transcription-polymerase chain reaction assay. J Immunol Methods. 1996;196:199–206. doi: 10.1016/0022-1759(96)00129-9. [DOI] [PubMed] [Google Scholar]
- 32.Freeman MR, Schneck FX, Gagnon Ml, et al. Peripheral blood T lymphocytes and lymphocytes infiltrating human cancers express vascular endothelial growth factor: a potential role for T cells in angiogenesis. Cancer Res. 1995;55:4140–5. [PubMed] [Google Scholar]
- 33.Cooper JC, Sharkey AM, McLaren J, et al. Localization of vascular endothelial growth factor and its receptor, flt, in human placenta and decidua by immunohistochemistry. J Reprod Fertil. 1995;105:205–13. doi: 10.1530/jrf.0.1050205. [DOI] [PubMed] [Google Scholar]
- 34.Brogi E, Tiangen W, Namiki A, Isner JM. Indirect angiogenic cytokines upregulate VEGF and bFGF gene expression in vascular smooth muscle cells, whereas hypoxia upregulates VEGF expression only. Circulation. 1994;90:649–52. doi: 10.1161/01.cir.90.2.649. [DOI] [PubMed] [Google Scholar]
- 35.Koochekpour S, Merzak A, Pilkington GJ. Vascular endothelial growth factor production is stimulated by gangliosides and TGF-beta isoforms in human glioma cells in vitro. Cancer Lett. 1996;102:209–15. doi: 10.1016/0304-3835(96)04161-4. [DOI] [PubMed] [Google Scholar]
- 36.Mandriota SJ, Menoud PA, Pepper MS. Transforming growth factor beta 1 down-regulates vascular endothelial growth factor receptor 2/flk-1 expression in vascular endothelial cells. J Biol Chem. 1996;271:115000–5. doi: 10.1074/jbc.271.19.11500. [DOI] [PubMed] [Google Scholar]
- 37.Liebovich SJ, Polverini PJ, Shepard HM, et al. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-α. Nature. 1987;329:630–2. doi: 10.1038/329630a0. [DOI] [PubMed] [Google Scholar]
- 38.Ryuto M, Ono M, Izumi H, et al. Induction of vascular endothelial growth factor by tumour necrosis factor alpha in human glioma cells. Possible roles of SP-1. J Biol Chem. 1996;271:28220–8. doi: 10.1074/jbc.271.45.28220. [DOI] [PubMed] [Google Scholar]
- 39.Elliott MJ, Maini RN, Feldmann M, et al. Randomised double-blind comparison of monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–10. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 40.Elliott MJ, Maini RN, Feldmann M, et al. Repeated therapy with monoclonal antibody to tumour necrosis factor alpha (cA2) in patients with rheumatoid arthritis. Lancet. 1994;344:1125–7. doi: 10.1016/s0140-6736(94)90632-7. [DOI] [PubMed] [Google Scholar]
- 41.Sunderkotter C, Steinbrink K, Goebeler M, et al. Macrophages and angiogenesis. J Leuk Biol. 1994;55:410–22. doi: 10.1002/jlb.55.3.410. [DOI] [PubMed] [Google Scholar]
- 42.Hart PH, Vitti GF, Burgess DR, et al. Potential anti-inflammatory effects of interleukin-4: suppression of human monocyte tumour necrosis factor α, interleukin-1 and prostaglandin F2. Proc Natl Acad Sci USA. 1989;86:3803–7. doi: 10.1073/pnas.86.10.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lacraz S, Nicod I, Galve-de Rochemonteux B, et al. Suppression of metalloproteinase biosynthesis in human alveolar macrophages by interleukin-4. J Clin Invest. 1992;90:382–6. doi: 10.1172/JCI115872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katsikis P, Chu CQ, Brennan FM, et al. Immunoregulatory role of interleukin 10 (IL-10) in rheumatoid arthritis. J Exp Med. 1994;179:1517–27. doi: 10.1084/jem.179.5.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Llorente L, Richaud-Patin Y, Fior R, et al. In vivo production of interleukin-10 by non-T cells in rheumatoid arthritis, Sjogrens syndrome, and systemic lupus erythematosus. Arthritis Rheum. 1994;37:1647–55. doi: 10.1002/art.1780371114. [DOI] [PubMed] [Google Scholar]
- 46.Hopkins SJ, Meager A. Cytokines in synovial fluid II. The presence of tumour necrosis factor and interferon. Clin Exp Immunol. 1988;73:88–92. [PMC free article] [PubMed] [Google Scholar]
- 47.Di Giovine FS, Nuki G, Duff GW. Tumour necrosis factor in synovial exudates. Ann Rheum Dis. 1998;47:768–72. doi: 10.1136/ard.47.9.768. [DOI] [PMC free article] [PubMed] [Google Scholar]