

Abstract
The NOD mouse has been used to explore the many features of insulin-dependent diabetes mellitus (IDDM) that is caused by the destruction of insulin-producing β cells in the islets of Langerhans of the pancreas. Self-reactive T cells have been considered to mediate IDDM in the NOD mouse, and antigen-presenting cells like DC and macrophages are expected to be involved in the processes from their role in generating regulatory or effector T cells. The present study shows that transfer of IFN-γ-stimulated DC of the NOD or ICR mouse into the NOD mouse did not accelerate IDDM onset but afforded long-lasting protection against clinical and histological signs of IDDM in the recipient mice. The anti-diabetogenic ability was unique to IFN-γ-stimulated DC when compared with unstimulated DC. A considerable proportion of the injected IFN-γ-stimulated DC was demonstrated to migrate into the pancreas and its associated lymphoid tissues, suggesting the DC exert their anti-diabetogenic effects there. These findings suggest that development of autoimmune diabetes in the NOD mouse is under the control of DC, and that IDDM onset could be controlled by appropriately manipulating DC systems in vivo, which may open the gate for the therapeutic application of ex vivo-conditioned DC to human IDDM.
Keywords: insulin-dependent diabetes mellitus, insulitis, antigen-presenting cells, adoptive transfer
INTRODUCTION
Insulin-dependent diabetes mellitus (IDDM) is caused by the destruction of insulin-producing β cells in the islets of Langerhans of the pancreas [1–3]. The NOD mouse has been used to explore IDDM and its relation to human IDDM: a NOD mouse develops a characteristic autoimmune lesion in the islets of Langerhans with lymphocytic infiltration—first by DC and macrophages and then by T cells (CD4+ and CD8+) and B cells—and destruction of pancreatic β cells follows [4–7]. The results are hypoinsulinaemia, hyperglycaemia, ketoacidosis and death, as observed in humans.
Considerable evidence suggests that IDDM in the NOD mouse is mediated by self-reactive T cells [8–11]. Thus, antigen-presenting cells (APC), like DC and macrophages, are expected to be involved because of their role in generating regulatory or effector T cells. Among various APC, DC are known to be professional APC that are well equipped for activation of naive T cells, to play a crucial role in controlling immune responses, and to either augment or reduce autoimmune responses by a variety of mechanisms [12–14]. Indeed, DC are an early component of the islet infiltration in the NOD mouse [6, 7], where DC may acquire relevant antigens and initiate an immune response, leading to β cell destruction by presenting the antigens to T cells. T cells from the NOD mouse display an abnormally high reactivity to self proteins [15]. However, when NOD mice were injected with immunopotentiators such as live bacille Calmette–Guérin (BCG) or Freund's complete adjuvant (FCA) containing a mycobacterial cell wall, whose primary targets appeared to be macrophages and DC, IDDM was almost completely prevented in the mice [16, 17]. These findings suggest that development of autoimmune diabetes in the NOD mouse is under the control of DC. In other words, IDDM onset could be controlled by appropriately manipulating DC systems in vivo. In the present study, it is shown for the first time that insulitis and IDDM in the NOD mouse can largely be prevented by transfer of DC stimulated ex vivo with IFN-γ.
MATERIALS AND METHODS
Animals
NOD and ICR mice were obtained from Clea Japan Inc. (Tokyo, Japan), and bred in a specific pathogen-free animal facility of Ehime University School of Medicine. In this sample of NOD mice, insulitis occurred at the age of 4–5 weeks, and diabetes spontaneously developed in about 70% of the female mice between 12 and 30 weeks of age. Donors of DC were 8–12-week-old non-diabetic female NOD or ICR mice. Recipients of the DC transfer were 1 or 4-week-old NOD female mice. Littermates were randomly divided into various experimental groups to control for potential variation in the incidence of diabetes among different litters.
Monitoring for diabetes
The urine of recipient NOD mice was monitored twice a week for glucose with Tes-Tape (Eli Lilly & Co., Indianapolis, IN) after transfer. Mice were defined as diabetic when they showed 2 consecutive days of > (+) glucosuria or their non-fasting blood glucose concentrations were > 200 mg/dl.
DC isolation
DC were prepared from the spleens of non-diabetic female NOD or ICR mice at the ages of 8–12 weeks according to the methods described previously with some modifications [18]. Splenic adherent cells consisting of DC and macrophages were prepared by culturing low-density cells from collagenase-digested spleens. After overnight culture, enriched populations of DC were recovered as non-adherent cells, depleted of Fc receptor-bearing cells by rosette formation with antibody-coated sheep erythrocytes, and further treated with a mixture of anti-Thy-1.2 (clone 5a-8; Cedarlane, Ontario, Canada), anti-Lyt-1.2 (clone CG16; Cedarlane) and anti-CD45R (clone RA3-3A1/6.1.TIB146; ATCC, Rockville MD) plus C to eliminate contaminating T and B cells, respectively.
Ex vivo treatment and transfer of DC
Purified DC (106 cells/ml) were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), 50 μm 2-mercaptoethanol (2-ME) and 20 μg/ml gentamycin in the presence or absence of 100 U/ml IFN-γ (specific activity 4.5–9 × 106 U/mg; Genzyme Co., Cambridge, MA) for 24 h. DC and IFN-γ-stimulated DC were washed, resuspended in PBS, and intraperitoneally transferred into 1- or 4-week-old female NOD mice (2 × 105 DC/200 μl per mouse). Control mice were intraperitoneally injected with 200 μl PBS. Survival curve analysis of diabetes incidence using the log rank test was performed to statistically analyse the data.
Histology
Pancreata of NOD mice were fixed in 10% formalin solution, mounted with paraffin and cut into 3-μm sections. The sections were stained with haematoxylin and eosin solution and observed using light microscopy. Insulitis was scored on at least 20 islets for each specimen by two independent, blinded observers according to the following criteria: grade 0, no lymphocytic infiltration; grade 1, lymphocytic infiltrations < 25% of the islet area; grade 2, lymphocytic infiltrations between 25% and 50% of the islet area; grade 3, lymphocytic infiltrations between 50% and 75%; grade 4, lymphocytic infiltrations > 75% of the islet area. The mean score for each pancreas was calculated by dividing the total score values by the number of islets scored. Immunohistochemical detection of insulin was performed using guinea pig anti-porcine insulin antibody (Nichirei Inc., Tokyo, Japan) together with biotin-conjugated anti-rabbit IgG (Nichirei) and peroxidase-conjugated streptavidin (Dako Co., Glostrup, Denmark).
DC migration study
The migration of DC, intraperitoneally transferred into NOD mice, was examined by labelling DC with 111In-chloride (Amersham Pharmacia Biotech Ltd, UK; RAC 370 mBq/ml, SA 1.85 GBq/μg) as previously described [19, 20]. In brief, 111In-chloride solution was mixed with tropolone (2-hydroxy-2,4,6-cyclohepatrienone; Sigma Chemical Co., St Louis, MO), and the 111In-tropolone was added to IFN-γ-stimulated DC suspension (approx. 1 μCi/106 cells) in RPMI 1640 medium containing 5% FCS and incubated for 5 min at 22°C. The DC were washed, resuspended in serum-free RPMI 1640 medium, and intraperitoneally injected into 4-week-old female NOD mice (2 × 106 cells/200 μl per mouse). The labelling efficiency was > 65%, and cell viability after labelling was > 90% as determined by the trypan blue dye exclusion test. At 24 h after cell transfer, the lung, liver, spleen, pancreas, gut and kidney were removed in their entirety for direct measurement of radioactivity in a well-type gamma counter. The activity in each tissue was expressed in two ways: (i) as a percentage of the total radioactivity injected; and (ii) as a percentage of the total radioactivity recovered per 0.1 g tissue sample (specific activity).
RESULTS
Prevention of diabetes onset by transfer of IFN-γ-stimulated DC
IFN-γ is well known to be one of the most powerful stimulators of DCmacrophages. In addition, in a small-scale pilot experiment, IFN-γ-treated DC showed anti-diabetogenic effect in the recipient NOD mouse among several reagents tested. We chose IFN-γ as a stimulant. DC were purified from spleens of 8–12-week-old NOD mice, treated with or without IFN-γ for 24 h and intraperitoneally transferred into 4-week-old NOD mice. The mice were then observed until the age of 30 weeks for the development of diabetes. The results of the experiments are shown in Fig. 1a. Control NOD mice that received injections of PBS developed diabetes in 24 of the 35 recipients (68.6%). NOD mice that had been given IFN-γ-stimulated DC from NOD mice developed diabetes in five of 19 recipients (26.3%), demonstrating that transfer of IFN-γ-stimulated DC significantly reduced diabetes onset in treated NOD mice (P < 0.01). In contrast, NOD mice given unstimulated DC from other NOD mice showed a slight reduction in developing diabetes (10/19, 52.6% diabetes), but it was not significant. Tests were conducted to determine whether DC from ICR mice, in which the NOD mouse strain was established, could prevent diabetes when transferred into NOD mice, since some functional defects were found in DC from NOD mice compared with those from ICR mice [21–23]. Contrary to expectations, however, diabetes in NOD mice was not significantly prevented by the transfer of DC from ICR mice (3/7, 42.8% diabetes), but was prevented by the transfer of DC stimulated with IFN-γ (2/8, 25.0% diabetes; P < 0.05), as shown in Fig. 1b.
Fig. 1.
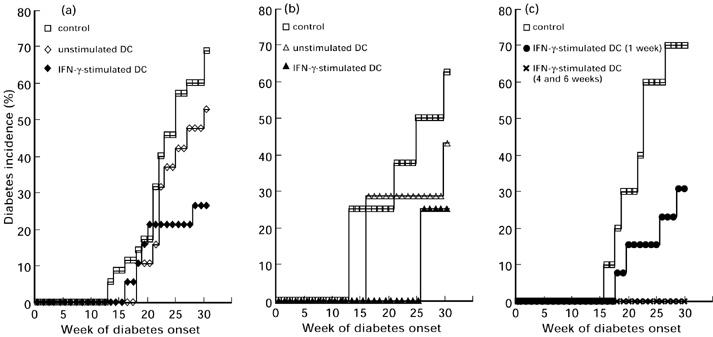
Prevention of diabetes onset by transfer of IFN-γ-stimulated DC. (a) DC purified from the spleen of NOD mice were stimulated with or without IFN-γ for 24 h and intraperitoneally transferred into 4-week-old NOD mice. Survival curve analysis of diabetes incidence using the log rank test was performed to analyse the data statistically. Control NOD mice developed diabetes in 24 of 35 recipients (68.6%) by the age of 30 weeks. NOD mice given IFN-γ-stimulated DC developed diabetes in five of 19 recipients (26.3%), and the reduction of the incidence was statistically significant (P < 0.01). Ten of 19 (52.6%) NOD mice given unstimulated DC developed diabetes; this was not significant. (b) DC purified from the spleen of ICR mice were stimulated with or without IFN-γ for 24 h and intraperitoneally transferred into 4-week-old NOD mice. Diabetes incidence of NOD mice given IFN-γ-stimulated DC was 2/8 (25.0%) (P < 0.05), and that of NOD mice given unstimulated DC was 3/7 (42.8%) (NS). (c) DC purified from the spleens of NOD mice were stimulated with IFN-γ for 24 h and intraperitoneally transferred into NOD mice once at 1 week old, or twice at 4 and 6 weeks old. The incidence of diabetes of NOD mice given IFN-γ-stimulated DC at 1 week old was 4/13 (30.8%) (P < 0.05), and that of NOD mice treated twice at 4 and 6 weeks old was 0/6 (0%) (P < 0.01).
Since it was found that transfer of DC from NOD or ICR mice equally prevented diabetes when they were ex vivo stimulated with IFN-γ, further studies were performed by using DC from NOD mice. We examined when and how recipient NOD mice were given IFN-γ-stimulated DC to prevent diabetes. When NOD mice were given IFN-γ-stimulated DC at 1 week old, the incidence of diabetes in the mice by the age of 30 weeks (4/13, 30.8% diabetes; P < 0.05; Fig. 1c) was about the same as that in mice that were given IFN-γ-stimulated DC at 4 weeks of age (Fig. 1a). Yet when NOD mice were given IFN-γ-stimulated DC at 6 weeks old, the anti-diabetogenic effect of the transfer was not always observed (data not shown), indicating that there is a critical period during the early growth of NOD mice for developing diabetes later. Furthermore, additional transfer of IFN-γ-stimulated DC was found to strengthen the effect of the first transfer, as shown in Fig. 1c: that is, diabetes was completely prevented in NOD mice that had received IFN-γ-stimulated DC twice, at 4 and 6 weeks of age (0/6, 0% diabetes; P < 0.01).
Pancreatic histology of NOD mice
The effect of IFN-γ-stimulated DC transfer on the insulitis process of NOD mice was assessed by histological examination of the pancreas. The pancreata of NOD mice that received IFN-γ-stimulated DC at 1 or 4 weeks of age and those from the control NOD mice that received only PBS at the same ages were microscopically examined and evaluated at 10 or 25 weeks old. As shown in Fig. 2, insulitis scores of 10-week-old NOD mice were not high regardless of the IFN-γ-stimulated DC transfer. The insulitis score of 25-week-old control NOD mice increased to 3.28, while those of NOD mice that received IFN-γ-stimulated DC at the ages of 1 week or 4 weeks were 0.47 (P < 0.01) and 1.00 (P < 0.01), respectively. Blood glucose levels of the three groups of mice were 97.2 mg/dl, 94.6 mg/dl and 95.4 mg/dl, respectively. These results indicate that the transfer of IFN-γ-stimulated DC prevents the histological changes in the pancreata of NOD mice that inevitably precede diabetes onset. Further, we immunohistochemically detected insulin-producing β cells in the pancreata of NOD mice. As shown in Fig. 3, the pancreata of 25-week-old control NOD mice were infiltrated with many mononuclear leucocytes and β cells had largely disappeared (Fig. 3A,B). In contrast, the infiltration of mononuclear leucocytes was not so apparent in the pancreas of IFN-γ-stimulated DC-transferred NOD mice, where many β cells were retained (Fig. 3C,D).
Fig. 2.
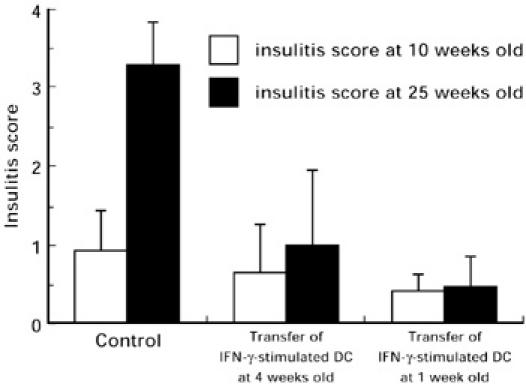
Prevention of insulitis by transfer of IFN-γ-stimulated DC. Pancreata of NOD mice when 10 and 25 weeks old that had received IFN-γ-stimulated DC at l week or 4 weeks old and those of control NOD mice were microscopically evaluated for the intensity of insulitis, and the scores were assessed as described in Materials and Methods. Differences between the insulitis score at 25 weeks old of the control group and those of the IFN-γ-stimulated DC transfer groups were significant by Student’s t-test (P < 0.01, P < 0.01, respectively).
Fig. 3.

Histological analysis of pancreata in NOD mice given IFN-γ-stimulated DC. Sections prepared from the pancreata of 25-week-old control NOD mice (A and B, grade 4; mag. × 100) and 25-week-old NOD mice that had been given IFN-γ-stimulated DC at 4 weeks old (C and D, grade 0; mag. × 80) were stained with haematoxylin and eosin (A,C) or immunohistochemically examined for insulin-producing β cells (B,D).
Tissue distribution of DC transferred into NOD mice
To assess the distribution of DC in the recipient NOD mice, we labelled IFN-γ-stimulated DC with 111In-chloride and transferred them into the peritoneal cavity of 4-week-old NOD mice. After 24 h, the lung, liver, spleen, pancreas, gut and kidney were removed in their entirety and their radioactivity was measured. About 20% of the radioactivity was distributed in these organs. The percentages of this organ radioactivity to total input radioactivity are shown in Fig. 4a. Percent radioactivity per 0.1 g of tissue was calculated and is shown in Fig. 4b. These values indicate that the pancreas and spleen showed the most radioactivity based on mass. This finding indicates that transferred DC migrate into the pancreas and pancreas-associated lymphoid tissues.
Fig. 4.
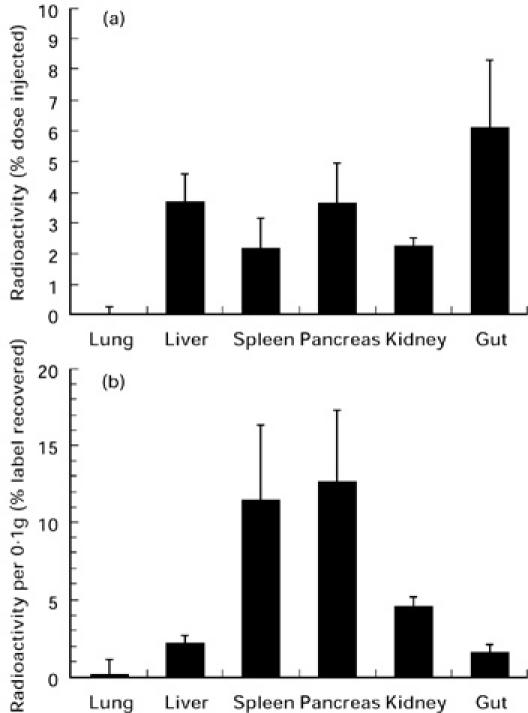
Tissue distribution of DC transferred into NOD mice. Purified DC (2 × 106 cells) were stimulated with IFN-γ, labelled with 111In-tropolone and intraperitoneally injected into 4-week-old NOD female mice. After 24 h, the lung, liver, spleen, pancreas, gut and kidney were removed in their entirety and their radioactivity was measured. The radioactivity in each organ is expressed as a percentage of the total radioactivities injected (a), and as a percentage of the total radioactivity recovered per 0.1 g tissue (b).
DISCUSSION
The present study for the first time shows that transfer of IFN-γ-stimulated DC into NOD mice did not accelerate IDDM onset but afforded long-lasting protection against clinical and histological signs of IDDM in the recipient mice. The anti-diabetogenic ability was unique to IFN-γ-stimulated DC compared with unstimulated DC. It had been expected that the transfer of DC from ICR mice, from which the NOD strain of mice was derived, would prevent diabetes in NOD mice because the ICR mice usually did not develop IDDM. However, even in this case, ex vivo treatment with IFN-γ was essential for DC to exert anti-diabetogenic effects. Based on mass a significant proportion of the injected IFN-γ-stimulated DC was demonstrated to migrate into the pancreas and associated lymphoid tissues, suggesting that the DC exert their diabetes-preventive effects there.
How can these data be reconciled with the general view that autoimmune diseases are initiated by autoreactive T cell responses toward self-antigens presented by DC? Indeed, T cells from the NOD mice displayed an abnormally high reactivity to self-proteins [15]. Thus, NOD mice which are deficient in CD4+ T cells do not develop insulitis [9], transfer of CD4+ islet-specific T cells into NOD mice accelerates IDDM onset [10], and antibodies against T cell surface molecules involved in T cell recognition suppress the development of insulitis in NOD mice [11]. DC have the ability to bind, process, and present antigens, including self-antigens [12, 13]. Presentation by such potent APC, well equipped for activation of naive CD4+ T cells, is usually considered to be immunogenic [24, 25]. DC are further stimulated to up-regulate costimulatory molecules that induce optimal activation of unprimed T cells [26]. Moreover, NOD mice possess the unusual H-2g7 MHC haplotype that is thought to encode the major component of IDDM susceptibility [27], and their APC are unable to activate tolerogenic mechanisms but remain capable of activating effector responses [21, 28]. Therefore it could be conceived that the transfer of IFN-γ-stimulated DC into the NOD mice would promote the development of autoimmune diabetes. Yet experimental results obtained in this study contradict this logic.
Nevertheless, recently there have been several reports that seem to support these findings. Clare-Salzler et al. reported that the transfer of DC of pancreatic lymph nodes from the NOD mouse prevented IDDM onset instead of accelerating the disease [29]. The authors postulated that the DC from pancreatic lymph nodes captured and presented some self islet antigens. Although DC were not further stimulated ex vivo with IFN-γ in their experiments, it is highly likely that the DC from the pancreatic lymph nodes were already exposed in the lymph nodes to IFN-γ secreted by T cells which recognized and responded to islet antigens, because DC obtained from other sites did not show the ability to prevent IDDM in their experiment, just like unstimulated DC in the present study. We reason that both the DC from pancreatic lymph nodes and the ex vivo IFN-γ-stimulated DC prevent IDDM in the NOD mouse in a similar manner. Another report showed that administration of IFN-γ, contrary to the authors' expectations, prevented IDDM in DP-BB rats, which is very similar to IDDM in NOD mice [30]. IFN-γ is the cytokine that can inhibit Th2 cell differentiation and effector functions, and lead to a dominant Th1 response that is generally believed to provoke and aggravate autoimmune diseases [31]. Furthermore, FCA, which is a powerful stimulator of DC or macrophages and commonly used for the enhancement of immune responses, was reported paradoxically to prevent IDDM in NOD mice [17], whereas mice must be immunized with autoantigens emulsified in FCA to induce experimental autoimmune diseases [32]. These past findings, combined with this study's results, lead to speculation that DC stimulated with IFN-γ or other stimulants do not immediately trigger but somehow control autoimmune responses in the NOD mouse. In this regard, it is of great interest that there has been increasing evidence demonstrating that DC, in some circumstances, can switch off immune responses instead of activating them [33–35]; the DC may induce tolerance or negatively regulate immune responses.
These reports have enabled us to postulate that the transfer of IFN-γ-stimulated DC down-regulates the autoimmune response, leading to IDDM in the NOD mouse. The mechanisms by which the transferred DC negatively regulate autoimmune responses in the NOD mouse have not yet been clarified, but there seem to be two critical points: the age of the recipient NOD mouse and the treatment of DC with IFN-γ. When DC were transferred to the NOD mouse older than 6 weeks, the anti-diabetogenic effect was not remarkable, suggesting that the transferred DC can exert their anti-diabetogenic effect before the self-reactive T cell population is established in the NOD mouse. It is essential for the DC to be treated with IFN-γ in advance of the adoptive transfer to acquire anti-diabetogenic activity, since untreated DC of the NOD or the ICR mouse did not prevent IDDM even when they were transferred to the NOD mouse at the age of 4 weeks. It was also demonstrated that a considerable proportion of the transferred DC migrated into the pancreas and its associated lymphoid tissues, suggesting the DC can function there. The pattern of distribution is thought to be dependent on the route of transfer. Thus, the route of DC transfer seems to be another important factor. Taking these into consideration, we think that the transferred IFN-γ-stimulated DC prevent IDDM by negatively controlling the generation of T cells that attack self-antigens in the islets of the pancreas. How can DC be changed by stimulation with IFN-γ? We have recently found that stimulation of DC with IFN-γ leads to inducing the expression of more than 30 distinct genes (M. Shinomiya, unpublished observation), though it has not yet been determined which of the genes are responsible for the prevention of IDDM in the recipient NOD mouse. Experiments are currently being performed to clarify this point.
From the clinical point of view [36], ex vivo IFN-γ-stimulated DC are thought to have an advantage over DC from pancreatic lymph nodes in the yield of cells. The DC in pancreatic lymph nodes need to be pulsed with some self-antigens first to stimulate self-reactive T cells to produce cytokines like IFN-γ, and then the cytokines stimulate the DC. A large yield of DC from these particular lymph nodes cannot be expected. In contrast, generation of the DC having an anti-diabetogenic capacity by directly incubating them with IFN-γ does not require the lymph node apparatus. Those who are predisposed to IDDM can in many cases be diagnosed before disease onset [37, 38]. The method by which human mononuclear cells are isolated in large quantities using the combination of leukopheresis and centrifugal elutriation, stimulated ex vivo with IFN-γ, and returned to patients under germ-free conditions, has recently been established [39]. Clinical trials using ex vivo-treated DC to human IDDM are being designed in this clinic.
Acknowledgments
The authors would like to express their gratitude to all involved in this study, especially to Dr Kayo Inaba (Department of Zoology, Kyoto Unversity, Kyoto, Japan) for helpful discussions.
REFERENCES
- 1.Bach JF. Insulin-dependent diabetes mellitus. Curr Opin Immunol. 1991;3:902–5. doi: 10.1016/s0952-7915(05)80011-1. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson MA, Maclare NK. The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med. 1994;331:1428–36. doi: 10.1056/NEJM199411243312107. [DOI] [PubMed] [Google Scholar]
- 3.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–7. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 4.Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and related strains. Adv Immunol. 1992;51:285–322. doi: 10.1016/s0065-2776(08)60490-3. [DOI] [PubMed] [Google Scholar]
- 5.Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7:727–38. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- 6.O'Reilly LA, Huchings PR, Crocker PR, et al. Characterization of pancreatic islet cell infiltration in NOD mice: effect of cell transfer and transgene expression. Eur J Immunol. 1991;21:1171–80. doi: 10.1002/eji.1830210512. [DOI] [PubMed] [Google Scholar]
- 7.Jansen AF, Homo-Delarche H, Hooijkaas PJ, Leenen PJ, Dardenne M, Drexhage HA. Immunohistochemical characterization of monocytes-macrophages and dendritic cells involved in the initiation of the insulitis and β-cell destruction in NOD mice. Diabetes. 1994;43:667–75. doi: 10.2337/diab.43.5.667. [DOI] [PubMed] [Google Scholar]
- 8.Miyazaki A, Hanafusa T, Yamada K, et al. Predominance of T lymphocytes in pancreatic islets and spleen of pre-diabetic non-obese diabetic (NOD) mice: a longitudinal study. Clin Exp Immunol. 1985;60:622–30. [PMC free article] [PubMed] [Google Scholar]
- 9.Bendelac A, Carnaud C, Boitard C, Bach JF. Synergistic transfer of autoimmune diabetes from L3T4+ and Lyt-2+ T cells. J Exp Med. 1987;166:823–32. doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+ islet-specific T cell clone. Science. 1990;249:1433–6. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 11.Semoe P, Bedossa P, Richard MF, Villa MC, Bach JF, Boitard C. Anti-alpha/beta T cell receptor monoclonal antibody provides an efficient therapy for autoimmune diabetes in nonobese diabetic (NOD) mice. Eur J Immunol. 1991;21:1163–9. doi: 10.1002/eji.1830210511. [DOI] [PubMed] [Google Scholar]
- 12.Hart DNJ. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90:3245–87. [PubMed] [Google Scholar]
- 13.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 14.Knight SC, Farrant J, Chan J, Bryant H, Bedford PH, Bateman C. Induction of autoimmunity with dendritic cells: studies on thyroiditis in mice. Clin Immunol Immunopathol. 1988;48:277–89. doi: 10.1016/0090-1229(88)90021-9. [DOI] [PubMed] [Google Scholar]
- 15.Ridgway WM, Fasso M, Lanctot A, Garvey C, Fathman CG. Breaking self-tolerance in nonobese diabetic mice. J Exp Med. 1996;183:1657–62. doi: 10.1084/jem.183.4.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harada M, Kishimoto Y, Makino S. Prevention of overt diabetes and insulitis in NOD mice by a single BCG vaccination. Diabetes Res Clin Pract. 1990;8:85–89. doi: 10.1016/0168-8227(90)90017-n. [DOI] [PubMed] [Google Scholar]
- 17.Ulaeto D, Lacy PE, Kipnis DM, Kanagawa O, Unanue ER. A T-cell dormant state in the autoimmune process of nonobese diabetic mice treated with complete Freund's adjuvant. Proc Natl Acad Sci USA. 1992;89:3927–31. doi: 10.1073/pnas.89.9.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crowley M, Inaba K, Witmar-Pack M, Steinman RM. The cell surface of mouse dendritic cells from different tissue including thymus. Cell Immunol. 1989;118:108–25. doi: 10.1016/0008-8749(89)90361-4. [DOI] [PubMed] [Google Scholar]
- 19.Kupiec-Weglinski JW, Austin JM, Morris PJ. Migration patterns of dendritic cells in the mouse. Traffic from the blood, and T cell-dependent and -independent entry to lymphoid tissues. J Exp Med. 1988;167:632–45. doi: 10.1084/jem.167.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dewanjee MK, Rao SA, Didisheim P. Indium-111 tropolone, a new high-affinity platelet label: preparation and evaluation of labeling parameters. J Nucl Med. 1981;22:981–7. [PubMed] [Google Scholar]
- 21.Serreze DV, Gaskins HR, Leiter EH. Defects in the differentiation and function of antigen presenting cells in NOD/Lt mice. J Immunol. 1993;150:2534–43. [PubMed] [Google Scholar]
- 22.Serreze DV, Gaedeke JW, Leiter EH. Hematopoietic stem-cell defects underlying abnormal macrophage development and maturation in NOD/Lt mice: defective regulation of cytokine receptors and protein kinase C. Proc Natl Acad Sci USA. 1993;90:9625–9. doi: 10.1073/pnas.90.20.9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jansen A, van Hagen M, Drexhage HA. Defective maturation and function of antigen-presenting cells in type 1 diabetes. Lancet. 1995;345:491–2. doi: 10.1016/s0140-6736(95)90586-3. [DOI] [PubMed] [Google Scholar]
- 24.Guery JC, Adorini L. Dendritic cells are the most efficient in presenting endogenous naturally processed self-epitopes to class II-restricted T cells. J Immunol. 1995;154:536–44. [PubMed] [Google Scholar]
- 25.Ranjeny R, Lipsky PE. Could endogenous self-peptides presented by dendritic cells initiate rheumatoid arthritis? Immunol Today. 1996;17:559–64. doi: 10.1016/s0167-5699(96)20030-1. [DOI] [PubMed] [Google Scholar]
- 26.Larsen CP, Ritchie SC, Hendrix R, et al. Regulation of immunostimulatory function and costimulatory molecule (B7-1 and B7-2) expression on murine dendritic cells. J Immunol. 1994;152:5208–19. [PubMed] [Google Scholar]
- 27.Wicker LS, Miller BJ, Coker LZ, et al. Genetic control of diabetes and insulitis in the non-obese diabetic (NOD) mouse. J Exp Med. 1987;165:1639–54. doi: 10.1084/jem.165.6.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrasco-Marin E, Shimizu J, Kanagawa O, Unanue ER. The Class II MHC I-Ag7 molecules from non-obese diabetic mice are poor peptide binders. J Immunol. 1996;156:450–8. [PubMed] [Google Scholar]
- 29.Clare-Salzler MJ, Brooks J, Chai A, Herle KV, Anderson C. Prevention of diabetes in nonobese diabetic mice by dendritic cell transfer. J Clin Invest. 1992;90:741–8. doi: 10.1172/JCI115946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicoletti F, Zaccone P, Marco RD, et al. Paradoxical antidiabetogenic effect of γ-interferon in DP-BB rats. Diabetes. 1998;47:32–38. doi: 10.2337/diab.47.1.32. [DOI] [PubMed] [Google Scholar]
- 31.Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;16:34–38. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 32.Hinrichs DJ, Wegmann KW, Dietsch GN. Transfer of experimental allergic encephalomyelitis to bone marrow chimeras. J Exp Med. 1987;166:1906–11. doi: 10.1084/jem.166.6.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farrant J, Bryant AE, Chan J, Hinsworth RL. Thyroglobulin treated blood dendritic cells reduce IgG anti-thyroglobulin antibody in vitro in Hashimoto's thyroiditis. Immunol Immunopathol. 1986;41:433–41. doi: 10.1016/0090-1229(86)90014-0. [DOI] [PubMed] [Google Scholar]
- 34.Finkelman FD, Lees A, Birnbaum R, Gause WC, Morris SC. Dendritic cells can present antigen in vivo in a tolerogenic or immunogenic fashion. J Immunol. 1996;157:1406–14. [PubMed] [Google Scholar]
- 35.Suss G, Shortman K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J Exp Med. 1996;183:1789–96. doi: 10.1084/jem.183.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giampiero G, Ricciardi-Castagnoli P. Dendritic cells hold promise for immunotherapy. Immunol Today. 1997;18:102–4. doi: 10.1016/s0167-5699(97)01030-x. [DOI] [PubMed] [Google Scholar]
- 37.Verge CF, Gianani R, Kawasaki E, et al. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 1996;45:926–33. doi: 10.2337/diab.45.7.926. [DOI] [PubMed] [Google Scholar]
- 38.Totta F, Gianani R, Previti M, et al. Autoimmunity to the GM2-1 islet ganglioside before and at the onset of type I diabetes. Diabetes. 1996;45:1193–6. doi: 10.2337/diab.45.9.1193. [DOI] [PubMed] [Google Scholar]
- 39.Shinomiya H, Shinomiya M, Stevenson GW, Stevenson HC. Activated killer monocytes: preclinical model systems. In: Stevenson HC, editor. Adoptive cellular immunotherapy of cancer (Immunology Series 48) New York: Marcel Dekker; 1989. pp. 127–38. [PubMed] [Google Scholar]