

Abstract
ANCA, implicated as having a pathogenic role in systemic vasculitis, can activate tumour necrosis factor-alpha (TNF-α)-primed neutrophils by cross-linking surface-expressed ANCA antigens with neutrophil FcγRIIa receptors to release reactive oxygen species. The FcγRIIa receptor exists as polymorphic variants, R131 and H131, which differ in their ability to ligate human IgG2 and IgG3. Neutrophils homozygous for the FcγRIIa-H131 allotype bind more efficiently to IgG3 than the FcγRIIa-R131 allotype and are the only human FcγR which bind IgG2. Our aim was to determine whether the homozygous FcγRIIa-H131 individuals are more susceptible to developing ANCA-associated systemic vasculitis and nephritis due to differential IgG binding and activation. FcγRIIa allotype was determined by both allele-specific polymerase chain reaction (PCR) and Southern blotting with allele-specific oligonucleotide probes end-labelled with 32P-γATP, after PCR amplification of genomic FcγRIIa DNA in 107 Caucasian patients with ANCA+ vasculitis (of whom 89 had renal disease) and 100 ethnically matched controls. Phenotyping of neutrophil FcγRIIa alleles was confirmed in some patients by quantitative flow cytometry using murine MoAbs 41H16 and IV.3. Of the patients with ANCA+ systemic vasculitis, 75 had ANCA with specificity for proteinase 3 and 32 with specificity for myeloperoxidase. Overall, no skewing in FcγRIIa allotypes was seen in patients compared with controls. No significant increase of the FcγRIIa-H131 allotype was found amongst patients irrespective of ANCA specificity, and no association between the FcγRIIa allotype and nephritis was found. Our data suggest that the FcγRIIa receptor allotype is not a major factor predisposing to the development of ANCA+ systemic vasculitis, or to nephritis.
Keywords: FcγRIIa receptor, polymorphism, ANCA, systemic vasculitis
INTRODUCTION
Neutrophils play a key role in mediating the inflammatory vascular lesions associated with systemic vasculitis. The inflammatory lesions found in systemic vasculitis are typically neutrophil-rich [1, 2], and there is a strong correlation between the extent of renal involvement and the number of neutrophils present in renal biopsies [1, 2]. ANCA are present in the sera of patients with systemic vasculitis and bind to the antigens proteinase 3 (PR3) or myeloperoxidase (MPO) expressed on the surface of primed neutrophils, leading to their activation [3–6]. The mechanisms underlying neutrophil activation by ANCA are only partially elucidated. Falk et al. were the first to demonstrate that ANCA can activate tumour necrosis factor-alpha (TNF-α)-primed neutrophils to release superoxide anions and lysosomal enzymes [7]. Subsequent studies showed that simultaneous binding of neutrophil ANCA antigens and FcγIIa receptors by ANCA are necessary for the activation process [8–10]. Whole ANCA IgG was able to trigger superoxide anion production, whereas F(ab′)2 fragments of human ANCA or IgM murine ANCA failed to do so [9]. Although some investigators were able to induce a respiratory burst from neutrophils using ANCA F(ab′)2 [7, 11, 12], involvement of FcγRIIa receptors is supported by the finding that Fab fragments of anti-FcγRIIa receptor antibody could reduce by 23–80% the respiratory burst from human neutrophils, stimulated with murine and human-derived ANCA IgG [9, 10, 12, 13].
FcγRIIa receptors exhibit a genetically determined polymorphism (FcγRIIa-R131 and FcγRIIa-H131), resulting in differential ability to recognize and be activated by human and murine IgG isotypes. This polymorphism results from a single base substitution from guanidine to adenine at nucleotide 494 of the coding region in exon 4 [14–16], which leads to an amino acid change from arginine to histidine at position 131 of the second extracellular domain [17–20]. The H131 allele (494A) codes for histidine at position 131 and has a high affinity for human IgG2 and a low affinity for murine IgG1, whilst the R131 allele (494G) has arginine at position 131 and has little or no affinity for human IgG2 but has a high affinity for murine IgG1 [17–22]. To a lesser extent, the two allotypes also differ in their ability to ligate human IgG3, with the homozygous FcγRIIa-H131 allotype having the higher affinity for IgG3 [17–22]. Thus, this polymorphism may influence susceptibility to diseases, especially in situations where IgG2, and to a lesser extent IgG3, are the predominant antibody subclasses produced. Salmon et al. have shown a reduction of the FcγRIIa-H/H131 genotype amongst patients with systemic lupus erythematosus [23]. This may represent more efficient binding and clearance of IgG2 immune complexes in individuals expressing the FcγRIIa-H/H131 allotype. Indeed, Haseley et al. found a predominance of IgG2 anti-C1q antibodies in patients with lupus, and the presence of FcγRIIa-R131 allele was associated with an increased risk of renal disease [24]. Duits et al. have also reported an increased prevalence of the FcγRIIa-R/R131 phenotype in lupus patients with renal disease compared with controls using indirect flow cytometry [25]. However, others have failed to find an association between FcγRIIa allotypes and the development of lupus [26].
This susceptibility to develop autoimmune disease and disease manifestations may be extended to include ANCA+ systemic vasculitis. Porges et al. have showed that the FcγRIIa allotype strongly influences the magnitude of the reactive oxygen burst by ANCA [10]. Homozygous FcγRIIa-R131 donors produced more than three-fold greater respiratory burst compared with the homozygous FcγRIIa-H131 donors in response to a murine monoclonal MPO-ANCA. However, this increased neutrophil activation in FcγRIIa-R131 donors almost certainly reflects the greater avidity of these receptors for murine IgG1 [10, 17–22]. The situation with human IgG is unclear. FcγRIIa receptor allotypes may represent risk factors in ANCA-associated systemic vasculitis, and influence susceptibility or disease manifestations if IgG2 and/or IgG3 are the predominant ANCA isotypes. MPO and PR3 are protein antigens, and isotypes IgG1 and IgG3 are produced in response to protein antigens [27]. Although ANCA activity can be found in all IgG subclasses, an over-representation of IgG3 is seen in one study with depletion of IgG2 in acute disease [28]. During remission, this subclass distribution reversed, with a fall in the IgG3 levels and an increase of IgG2 by approximately three-fold. In other studies, sera with relatively high levels of IgG3 subclass of ANCA can preferentially activate neutrophils [9], and renal exacerbations of Wegener's granulomatosis (WG) are associated with increases of the IgG3 subclass of ANCA [29]. Since the FcγRIIa-H131 receptor binds human IgG3 with greater avidity and also binds human IgG2, receptor engagement and antigen recognition by ANCA could lead to an enhanced neutrophil activation and tissue injury, compared with the FcγRIIa-R131 receptor. Thus allelic variants of neutrophil FcγR may contribute to disease susceptibility and organ involvement through differential activation by ANCA isotypes. To test the hypothesis that FcγRIIa alleles might influence susceptibility to the development of ANCA+ systemic vasculitis, we compared distribution of FcγRIIa allotypes in Caucasian patients with an ANCA+ vasculitis with that of healthy matched subjects. Specifically, the relationship between FcγRIIa allotype and nephritis was also examined.
SUBJECTS AND METHODS
Subjects for FcγRIIa allotyping
Peripheral blood was collected into tubes containing 1/10 volume of 0.5 m EDTA pH 8.0 from 100 disease-free individuals who attended hospital for minor surgery, and 107 patients with ANCA+ systemic vasculitis classified using the Chapel Hill Consensus Conference definitions [30]. WG was diagnosed if there was granulomatous inflammation of the respiratory tract, with or without involvement of other organs, and histological findings of necrotizing vasculitis affecting small to medium sized vessels. Churg–Strauss syndrome was diagnosed if there was evidence of eosinophil-rich and granulomatous inflammation involving the respiratory tract, necrotizing vasculitis affecting small to medium-sized vessels, asthma and blood eosinophilia. Microscopic polyangiitis was diagnosed if there was evidence of an inflammatory process affecting at least two organs (one of which was typically the kidneys) with histology showing necrotizing vasculitis with few or no immune deposits affecting small vessels. Polyarteritis nodosa was diagnosed if there was evidence of necrotizing inflammation of medium-sized or small arteries without glomerulonephritis. All patients and controls were Caucasians. Renal involvement was defined as a plasma creatinine of > 150 μmol/l, creatinine clearance of < 100 ml/min, the presence of haematuria, proteinuria > 0.5 g daily, or biopsy-proven histology showing necrotizing vasculitis.
Determination of FcγRIIa allotypes by allele-specific polymerase chain reaction
DNA extraction
Genomic DNA was extracted using a nucleic acid extraction kit (ORCA Research Inc., Bothwell, WA) either from peripheral blood obtained from subjects or from the cell lines K562 and U937. These cells had previously been shown to have the FcγRIIa-R/H131 and FcγRIIa-R/R131 allotypes, respectively [18, 31]. K562 and U937 cells were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS; Laboratory Technology Int., Stamborne, UK).
Polymerase chain reaction amplification
FcγRIIa DNA was amplified as described by Osborne et al. using gene-specific primers [32]: sense primer (5′-CAA GCC TCT GGT CAA GGT C-3′) and antisense primer (5′-GAA GAG CTG CCC ATG CTG-3′). The sense primer is upstream to the polymorphism encoding amino acid 131 in the second extracellular domain and does not distinguish between the genes for FcγRIIa, b or c. The antisense primer is located downstream of the polymorphism in the transmembrane domain and contains nine bases on the 3′ end of the primer which are unique to FcγRIIa gene.
Allele-specific polymerase chain reaction
FcγRIIa genotyping was accomplished by allele-specific polymerase chain reaction (PCR). Two more allele-specific PCR reactions were carried out on each PCR product obtained from the first round of PCR described above. Both reactions utilized a common antisense primer located downstream to the polymorphism (5′-CTA GCA GCT CAC CAC TCC TC-3′), and one of the two allele-specific primers. The two allele-specific primers were designed with the 3′ base complementary to the allovariable base (H131-specific: 5′-GAA AAT CCC AGA AAT TCT CCC A; R131-specific: GAA AAT CCC AGA AAT TCT CCC G-3′). The PCR reactions were performed in a total volume of 50 μl containing 1 μl of the first PCR product, 0.1 μm sense and antisense primers, 2.75 mm magnesium chloride, 5 μl of 10× reaction buffer (Promega, Southampton, UK), 200 μm deoxynucleotide triphosphates and 0.1 U Taq DNA polymerase (Promega). The cycle programme was set to the following parameters: 95°C for 5 min; 35 cycles of 95°C for 15 s, 58°C for 30 s, 72°C for 30 s, and a final step of 72°C for 10 min. The FcγRIIa-specific primers amplify a 290-bp product which was visualized by ethidium bromide staining in a 1% agarose gel dissolved in TAE buffer (Fig. 1).
Fig. 1.

Agarose gel electrophoresis of amplified DNA by polymerase chain reaction (PCR) using allele-specific primers for FcγRIIa-H/H131 and R/R131, as described in Subjects and Methods. The FcγRIIa-specific primers amplify a 290-bp product. Amplified DNA from nine individuals (three H/H131, three R/H131, and three R/R131) and two cell lines (K562, R/H131 and U937, R/R131). K, K562; U, U937.
Determination of FcγRIIa allotypes by Southern blotting
FcγRIIa genotyping was performed using PCR amplification of genomic DNA (as described above), followed by Southern blotting using allele-specific probes end-labelled with 32P-γATP as described by Osborne et al. [32]. The probes detect a single nucleotide difference (G or A) at base 494 which results in an arginine (R131 allele) or histidine (H131 allele) at amino acid 131 of the FcγRIIa protein. Representative Southern blots showing two superimposed blots which had been hybridized with the two allele-specific oligonucleotides are shown in Fig. 2.
Fig. 2.

Superimposed Southern blots of amplified DNA hybridized with 32P-γATP-labelled FcγRIIa allele-specific oligonucleotide probes (H/H131 and R/R131), as described in Subjects and Methods. Amplified DNA from nine individuals (three H/H131, three R/H131, and three R/R131) and two cell lines (K562, R/H131 and U937, R/R131). Neg, Negative; K, K562; U, U937.
Determination of FcγRIIa allotypes by flow cytometry
Neutrophil isolation
Neutrophils were isolated using a method adapted from Toothill et al. [33]. Peripheral blood was collected from healthy donors into tubes containing acid citrate dextrose (9:1 dilution). The blood cells were first sedimented across Hespan (2.5% hydroxyethyl starch), then neutrophils were isolated by density gradient centrifugation at 500 g using isotonic percoll. Neutrophils were 99% viable by trypan blue exclusion and were 98–99% pure when stained with haematoxylin.
Flow cytometry
Phenotyping of cells was determined for K562 cells, U937 cells, and neutrophils from eight healthy donors (three R/R131, two H/H131 and three R/H131) using quantitative flow cytometry using murine MoAbs 41H16 (a gift from Dr J. Van der Winkel, Utrecht, The Netherlands), which recognizes the R131 allele and IV.3 (Medarex, Annandale, NJ), which recognizes both the R131 and H131 alleles, as described by Gosselin et al. [34]. By comparing the relative fluorescence staining of 41H16 with that of IV.3, the cells could be separated into the homozygous FcγRIIa-R/R131, H/H131 or heterozygous R/H131 allotype. Homozygous FcγRIIa-R131 neutrophils have comparable amounts of 41H16 and IV.3 epitopes, and so the mean fluorescence of 41H16 should be similar to that of IV.3, giving a relative fluorescence ratio of > 0.8. For homozygous FcγRIIa-H131 neutrophils which only has the IV.3 epitope, the relative fluorescence ratio of 41H16/IV.3 should be < 0.2. Heterozygous FcγRIIa-R/H131 neutrophils should have approximately one half as many 41H16 sites as IV.3 sites, giving a relative fluorescence ratio of between 0.4 and 0.8. Results obtained by quantitative flow cytometry showing binding of 41H16 and IV.3 are shown in Fig. 3.
Fig. 3.

Expression of FcγRIIa receptors on human neutrophils with the (a) R/R131, (b) R/H131, and (c) H/H131 phenotypes. Each profile represents 10 000 neutrophils, stained with murine MoAbs IV.3 (binds both FcγRIIa alleles) and 41H16 (binds FcγRIIa-R131 only).
ANCA testing
Indirect immunofluorescence
ANCA activity of samples was determined by indirect immunofluorescence on ethanol-fixed neutrophils using standard techniques [6].
Antigen-specific ELISA
Ninety-six-well ELISA plates (Nunc, Roskilde, Denmark) were coated with the antigens MPO (1 μg/ml) (Calbiochem, Nottingham, UK) or PR3 (0.8 μg/ml; Genesis Diagnostics, Cambridge, UK) overnight at 4°C in 0.06 m sodium carbonate buffer pH 9.6. ELISA plates were washed four times between each of the following incubation steps with 0.15 m sodium chloride/0.05% Tween, and all subsequent incubations were conducted at 37°C. Test sera were diluted to 1:40 in 0.15 m Tris HCl/0.15 m sodium chloride/0.05% Tween-20/2% FCS (Laboratory Technology Int), then added to ELISA plates and incubated for 1 h. After washing, alkaline phosphatase-conjugated goat anti-human IgG (Sigma, Poole, UK) diluted 1:3000 in 0.15 m Tris HCl/0.15 m sodium chloride/0.05% Tween-20/2% FCS was added and incubated for 30 min. After washing, the substrate p-nitrophenyl phosphate (1 mg/ml) in 1 m diethanolamine, pH 9.6, was added. The reaction was terminated with 3 m sodium hydroxide after 10 min, and the plates read at 405 nm with a 690-nm reference filter. A value of 3 s.d. above the mean values of 100 ANCA− normal samples was taken as positive.
Statistical analysis
FcγRIIa allotypes and allele frequencies were analysed by applying the χ2 test (3 × 2 contingency table). To reject the null hypothesis, a probability of 0.05 (two-tailed) was used. We then analysed the FcγRIIa polymorphism as a dichotomous variable comparing the homozyygous FcγRIIa-H/H131 (the hypothesized at risk allotype) versus the FcγRIIa-R/R131 and FcγRIIa-R/H131 allotypes using the χ2 test in the controls and vasculitis subjects. Comparisons were made between ANCA+ vasculitis patients and matched controls in all cases.
RESULTS
Patient demographics
All patients were ANCA+ as determined by indirect immunofluorescence and antigen-specific ELISA. Seventy-five patients had PR3-ANCA and 32 had MPO-ANCA. Of the 107 ANCA+ vasculitis patients, 48 had WG (16 had the limited form), 54 patients had microscopic polyangiitis, four had classical polyarteritis nodosa and one patient had Churg–Strauss syndrome.
FcγRIIa genotype frequency
The FcγRIIa allotype results obtained by allele-specific PCR, Southern blotting technique and quantitative flow cytometry all concorded. The FcγRIIa genotype and allele frequency in all patients are shown in Table 1. No skewing was observed in the overall genotype distribution (χ2 = 0.1018, P = 0.95) or allele frequency (χ2 = 0.0059, P = 0.94) between ANCA+ vasculitis patients and healthy control subjects. To see whether FcγRIIa alleles were risk factors for the development of nephritis, the allotype frequency of patients with and without renal disease was examined. Altogether, 89 patients had renal involvement (as defined above) and 18 patients did not have renal involvement (Table 1). Again, no skewing was observed in the genotype distribution (χ2 = 0.0213, P = 0.99) or allele frequency (χ2 = 0.0003, P = 0.99) between those vasculitis patients with renal disease and healthy control subjects.
Table 1.
Distribution of FcγRIIa genotypes and allele frequencies in controls and vasculitis patients

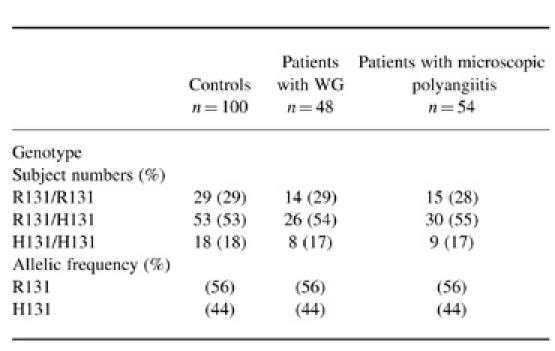
Forty-eight patients had WG and 54 patients had microscopic polyangiitis (Table 2). No skewing was observed in the overall genotype distribution (χ2 = 0.0414, P = 0.98) or allele frequency (χ2 = 0.000 01, P = 0.99) between patients with WG and healthy control subjects. Similarly, no skewing was observed in the overall genotype distribution (χ2 = 0.0964, P = 0.95) or allele frequency (χ2 = 0.0123, P = 0.91) between patients with microscopic polyangiitis and healthy control subjects
Table 2.
Distribution of FcγRIIa genotypes and allele frequencies in controls and patients with Wegener's granulomatosis (WG) and microscopic polyangiitis
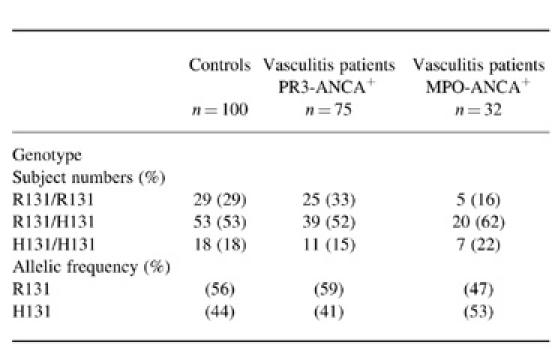
The genotype distribution and ANCA status are shown in Table 3. No skewing was observed in the overall genotype distribution (χ2 = 0.5563, P = 0.76) or allele frequency (χ2 = 0.3697, P = 0.54) between PR3-ANCA+ vasculitis patients and healthy control subjects. Similarly, no skewing was observed in the overall genotype distribution (χ2 = 2.2715, P = 0.32) or allele frequency (χ2 = 1.1236, P = 0.29) between MPO-ANCA+ vasculitis and healthy control subjects (χ2 = 2.2715, P = 0.32) and allele frequency (χ2 = 1.1236, P = 0.29). Specifically, when the frequency of FcγRIIa-H/H131, the hypothesized at risk genotype, was compared with FcγRIIa-R/R131 and FcγRIIa-R/H131, no significant increase was found in patients who were PR3-ANCA+ (χ2 = 0.1455, P = 0.70) or MPO-ANCA+ (χ2 = 0.0519, P = 0.82), compared with controls.
Table 3.
Distribution of FcγRIIa genotypes and allele frequencies in controls and ANCA+ patients
DISCUSSION
This study examines the hypothesis that FcγRIIa polymorphism may be a heritable factor influencing the ability of certain ANCA to initiate and promote inflammation, based on their relative binding affinities for the different IgG subclasses. The data indicate that FcγRIIa polymorphism is not a major factor predisposing to the development of ANCA-associated systemic vasculitis, since no skewing of the allotypes was found. In addition, no association with FcγRIIa allotypes and renal involvement was evident, which suggests that FcγRIIa alleles at least independently do not influence disease severity. Kimberly et al. using allele-specific PCR found the distribution of FcγRIIa allotypes in patients with WG to be indistinguishable from normal controls, as in our study [35]. In contrast, Piepot et al. using flow cytometry were able to show skewing towards the FcγRIIa-R/R131 allotype in patients with WG [36]. However, no significant difference in superoxide anion production was found when donor neutrophils with either homozygous FcγRIIa phenotype were stimulated with purified human ANCA [36]. This finding does not directly support a role for FcγRIIa polymorphism in ANCA-induced neutrophil activation that may underlie disease activity of systemic vasculitis. The current study utilized three methods to determine the FcγRIIa allotypes and included patients with other categories of ANCA-associated systemic vasculitis, aside from WG. The observations find no evidence to implicate a role for FcγRIIa polymorphism in the pathogenesis of systemic vasculitis. This conclusion is supported by studies of ANCA IgG subclasses. Although it has been suggested that IgG2 and IgG3 play a pathological role in ANCA+ vasculitis [9, 28, 29], IgG1 and IgG4 are present as well [29, 37–39]. Indeed, analysis of the subclasses of ANCA in sera from patients by a number of groups has established that IgG1 and IgG4 are the predominant subclasses found in active disease [29, 37–39]. It has been suggested by Porges et al. [10] that the under-representation of IgG2 compared with the other subclasses in the serum of patients with active vasculitis [28, 29, 37–39] may represent preferential neutrophil binding and clearance, especially in individuals with the FcγRIIa-H/H131 allotype. Indeed, Parren et al. found significantly lower IgG2 levels in non-vasculitic individuals expressing the IgG2-binding FcγRIIa-H/H131 allotypic form compared with the FcγRIIa-R/R131 allotypic form [19].
Despite the lack of influence of FcγRIIa polymorphism on the development of vasculitis, it remains possible that polymorphisms of other Fcγ receptors are important. Incomplete blocking of ANCA-mediated activation despite saturating doses of anti-FcγRIIa Fab, raises the possibility that there is also a non-FcγRIIa-dependent mechanism of cellular activation [28, 29, 37–39]. FcγRIIIb receptors are present on the surface of neutrophils and are involved in the activation of NADPH oxidase, a major component involved in the production of the respiratory burst [40, 41]. Like FcγRIIa, FcγRIIIb is heterogeneous and expresses allelic variants: NA1 and NA2 which encode proteins with differing capacities to trigger [42]. However, Porges et al. using quantitative flow cytometry failed to document any competition between ANCA which binds to FcγRIIa and MoAb 3G8 which recognizes the FcγRIIIb binding site [10]. This may reflect the rapid shedding and surface expression of this receptor and does not necessarily preclude FcγRIIIb receptor having a role in ANCA–neutrophil interaction. Indeed, Kocher et al. using flow cytometry were able to show engagement of neutrophil FcγRIIIb receptor by ANCA [43]. Engagement of the ligand binding site of FcγRIIIb by ANCA was assessed by competitive inhibition of a panel of anti-FcγRIII MoAbs and receptor shedding was blocked by the metalloproteinase inhibitor 1,10-phenanthroline. IgG ANCA inhibited the binding of anti-FcγRIII MoAbs 135.9 and 241.1, whilst MoAb 3G8, a high-affinity antibody capable of displacing other anti-CD16 MoAbs and most probably ligand, showed no change in binding. Thus FcγIIIb polymorphism might be important in disease pathogenesis in ANCA+ vasculitis by affecting quantitative receptor function. Recently using allele-specific PCR, Kimberly et al. characterized the distribution of FcγRIIIb alleles in 145 patients with WG [35]. The functionally more active NA1 allele of FcγRIIIb was significantly enriched in patients with renal disease. Polymorphisms of Fcγ may also act in combination with other, non-Fcγ polymorphisms to influence disease. For example, Fijen et al. found that individuals with a deficiency of a component of the terminal complement pathway (C6 or C8) in combination with both the FcγRIIa-R/R131 and the FcγRIIIb-NA2/NA2 allotypes had experienced more meningococcal infections than C6- or C8-deficient family members with other combinations of these FcγR allotypes [44]. In addition, co-operation between FcγRIIa and FcγRIIIb receptors has been described in a number of functional studies, including: the production of the respiratory burst [45], FcγRIIa-mediated phagocytosis [46], immune complex-induced neutrophil actin assembly [47], and the release of hydrolytic enzymes induced by IgM anti-FcγR autoantibodies [48]. These observations support an interaction between the magnitude of the humoral response and defined Fcγ receptor allotypes. Lastly, the possibility of a type II error can not be excluded in this study. In order to achieve an α value of 0.05 (two-sided) with 90% power, at least 600 subjects would be needed. It remains possible that FcγRIIa polymorphism acting synergistically with particular FcγRIIIb or other non-Fcγ polymorphisms may modulate disease susceptibility and expression in systemic vasculitis.
Acknowledgments
This work was funded by the Medical Research Council through a Training Fellowship for W.Y.T. Part of this work has been presented in abstract form at the Renal Association Meeting in London, UK (October 1996); 7th International ANCA Workshop in Rochester, Minnesota, USA (October 1996); and at the American Society of Nephrology in New Orleans, USA (November 1996). We thank Professor D. S. Kumararatne for his helpful discussions; Professor P. Bacon, for the provision of patients; Dr J. van de Winkel for the 41H16 antibody; and Dr K. Quibell for technical advice.
REFERENCES
- 1.Brouwer E, Huitema M, Mulder A, et al. Neutrophil activation in vitro and in vivo in Wegener's granulomatosis. Kidney Int. 1994;45:1120–31. doi: 10.1038/ki.1994.149. [DOI] [PubMed] [Google Scholar]
- 2.Adu D, Howie A. Vasculitis in the kidney. Curr Diagn Pathol. 1995;2:73–77. [Google Scholar]
- 3.Van der Woude F, Rasmussen N, Labatto S, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener's granulomatosis. Lancet. 1985;i:425–9. doi: 10.1016/s0140-6736(85)91147-x. [DOI] [PubMed] [Google Scholar]
- 4.Nolle B, Specks U, Ludemann J, Rohrbach M, DeRemee R, Gross W. Anti-cytoplasmic autoantibodies: their immunodiagnostic value in Wegener's granulomatosis. Ann Int Med. 1989;111:28–40. doi: 10.7326/0003-4819-111-1-28. [DOI] [PubMed] [Google Scholar]
- 5.Davies D, Moran J, Niall J, Ryan G. Segmental necrotizing glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? BMJ. 1982;285:606. doi: 10.1136/bmj.285.6342.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falk R, Jennette J. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med. 1988;318:1651–7. doi: 10.1056/NEJM198806233182504. [DOI] [PubMed] [Google Scholar]
- 7.Falk R, Terrell R, Charles L, Jennette J. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radical in vitro. Proc Natl Acad Sci USA. 1990;87:4115–9. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulder A, Horst G, Limburg P, Kallenberg C. Activation of neutrophils by anti-neutrophil cytoplasmic antibodies is FcR-dependent. Clin Exp Immunol. 1993;1(Suppl.):S16. [Google Scholar]
- 9.Mulder A, Heeringa P, Brouwer E, Limburg P, Kallenberg C. Activation of granulocytes by anti-neutrophil cytoplasmic antibodies (ANCA): a FcγRII-dependent process. Clin Exp Immunol. 1994;98:270–8. doi: 10.1111/j.1365-2249.1994.tb06137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porges A, Redecha P, Kimberly W, Csernok E, Gross W, Kimberly R. Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophils via FcγRIIa. J Immunol. 1994;153:1271–80. [PubMed] [Google Scholar]
- 11.Keogan M, Esnault V, Green A, Lockwood C, Brown D. Activation of normal neutrophils by anti-neutrophil cytoplasm antibodies. Clin Exp Immunol. 1992;90:228–34. doi: 10.1111/j.1365-2249.1992.tb07934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kettritz R, Jennette J, Falk R. Crosslinking of ANCA-antigens stimulates superoxide release by human neutrophils. J Am Soc Nephrol. 1997;8:386–94. doi: 10.1681/ASN.V83386. [DOI] [PubMed] [Google Scholar]
- 13.Reumaux D, Vossebeld P, Roos D, Verhoeven A. Effect of tumor necrosis factor-induced integrin activation on Fcγ receptor II-mediated signal transduction: relevance for activation of neutrophils by anti-proteinase 3 or anti-myeloperoxidase antibodies. Blood. 1995;86:3189–95. [PubMed] [Google Scholar]
- 14.Stuart S, Trounstine M, Vaux D, et al. Isolation and expression of cDNA clones encoding a human receptor for IgG (Fc-RII) J Exp Med. 1987;166:1668–84. doi: 10.1084/jem.166.6.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brooks D, Qiu W, Luster A, Ravetch J. Structure and expression of human IgG FcRII (CD32). Functional heterogeneity is encoded by the alternatively spliced products of multiple genes. J Exp Med. 1989;170:1369–85. doi: 10.1084/jem.170.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seki T. Identification of multiple isoforms of the low-affinity human IgG Fc receptor. Immunogenetics. 1989;30:5–12. doi: 10.1007/BF02421463. [DOI] [PubMed] [Google Scholar]
- 17.Clark M, Stuart S, Kimberly R, Ory P, Goldstein I. A single amino acid distinguishes the high responder form of Fc receptor II on human monocytes. Eur J Immunol. 1991;21:1911–6. doi: 10.1002/eji.1830210820. [DOI] [PubMed] [Google Scholar]
- 18.Warmerdam P, van de Winkel J, Ej G, Capel P. Molecular basis for a polymorphism of human Fcγ receptor II (CD32) J Exp Med. 1990;172:19–25. doi: 10.1084/jem.172.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parren P, Warmerdam P, Boeije L, et al. On the interaction of IgG subclasses with the low affinity FcγRIIa (CD32) on human monocytes, neutrophils, and platelets. Analysis of a functional polymorphism to human IgG2. J Clin Invest. 1992;90:1537–46. doi: 10.1172/JCI116022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tate B, Witort E, Mckenzie I, Hogarth P. Expression of the high responder/non-responder human FcγRII analysis by PCR and transfection into FcR-COS cells. Immunol Cell Biol. 1992;70:79–87. doi: 10.1038/icb.1992.12. [DOI] [PubMed] [Google Scholar]
- 21.Warmerdam P, van de Winkel J, Vlug A, Westerdaal N, Capel P. A single amino acid in the second Ig-like domain of the human Fcγ receptor II is critical for human IgG2 binding. J Immunol. 1991;147:1338–43. [PubMed] [Google Scholar]
- 22.Salmon J, Edberg J, Brogle N, Kimberly R. Allelic polymorphism of human Fcγ receptor IIa and Fcγ receptor IIIb. Independent mechanisms for differences in human phagocyte function. J Clin Invest. 1992;89:1274–81. doi: 10.1172/JCI115712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salmon J, Millard S, Schachter L, et al. FcγRIIa alleles are heritable risk factors for lupus nephritis. J Clin Invest. 1996;97:1348–54. doi: 10.1172/JCI118552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haseley LA, Wisnieski JJ, Denburg MR, et al. Antibodies to C1q in systemic lupus erythematosus: characteristics and relation to FcγRIIa alleles. Kidney Int. 1997;52:1375–80. doi: 10.1038/ki.1997.464. [DOI] [PubMed] [Google Scholar]
- 25.Duits A, Bootsma H, Derksen R, et al. Skewed distribution of IgG Fc receptor IIa (CD32) polymorphism is associated with renal disease in systemic lupus erythematosus patients. Arthritis Rheum. 1995;39:1832–6. doi: 10.1002/art.1780381217. [DOI] [PubMed] [Google Scholar]
- 26.Botto M, Theodoridis E, Thompson E, et al. FcγRIIa polymorphism in systemic lupus erythematosus (SLE): no association with disease. Clin Exp Immunol. 1996;104:264–8. doi: 10.1046/j.1365-2249.1996.33740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammerstrom L, Heigl Z, Smith C. IgG subclasses in bacterial infections. In: Shakib S, editor. Basic and clinical aspects of IgG subclasses. Basel: Karger; 1986. p. 134. [Google Scholar]
- 28.Jayne D, Weetman A, Lockwood C. IgG subclass distribution of autoantibodies to neutrophil cytoplasmic antigens in systemic vasculitis. Clin Exp Immunol. 1991;84:476–81. [PMC free article] [PubMed] [Google Scholar]
- 29.Brouwer E, Cohen Tervaert J, Horst G, et al. Predominance of IgG1 and IgG4 subclasses of anti-neutrophil cytoplasmic autoantibodies (ANCA) in patients with Wegener's granulomatosis and clinically related disorders. Clin Exp Immunol. 1991;83:379–86. doi: 10.1111/j.1365-2249.1991.tb05647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jennette J, Falk R, Andrassy K, et al. Nomenclature of systemic vasculitides: the proposal of an international consensus conference; Arthritis Rheum; 1994. pp. 187–92. [DOI] [PubMed] [Google Scholar]
- 31.Anderson C, Ryan D, Looney R, Leary P. Structural polymorphism of the human monocyte 40 kilodalton Fc receptor for IgG. J Immunol. 1987;138:2254–6. [PubMed] [Google Scholar]
- 32.Osborne J, Chacko G, Brandt J, Anderson C. Ethnic variation in frequency of an allelic polymorphism of human FcγRIIa determined with allele specific oligonucleotide probes. J Immunol Methods. 1994;173:207–17. doi: 10.1016/0022-1759(94)90299-2. [DOI] [PubMed] [Google Scholar]
- 33.Toothill V, Van-Mourik J, Niewenhuis H, Metzelaar M, Pearson J. Characterisation of the enhanced adhesion of neutrophil leukocytes to thrombin-stimulated endothelial cells. J Immunol. 1990;145:283–91. [PubMed] [Google Scholar]
- 34.Gosselin E, Brown M, Anderson C, Zipf T, Guyre P. The monoclonal antibody 41H16 detects the Leu 4 responder form of human FcγRII. J Immunol. 1990;144:1817–22. [PubMed] [Google Scholar]
- 35.Kimberly RP, Edberg JC, Wainstein E, et al. Association of the FcγRIIIb-NA1 allele with renal disease in Wegener's granulomatosis (WG) Clin Exp Immunol. 1998;112(Suppl. 1):26. [Google Scholar]
- 36.Piepot H, Heerings P, Cohen Tervaert J, Stegeman C, Duits A, Kallenberg C. FcγRIIa-polymorphism and neutrophil activation by anti-neutrophil cytoplasmic antibodies (ANCA) Clin Exp Immunol. 1995;101(Suppl. 1):35. [Google Scholar]
- 37.Esnault V, Jayne D, Weetman A, Lockwood C. IgG subclass distribution and relative functional affinity of anti-myeloperoxidase antibodies in systemic vasculitis at presentation and during follow-up. Immunol. 1991;74:714–8. [PMC free article] [PubMed] [Google Scholar]
- 38.Segelmark M, Wieslander J. IgG subclasses of antineutrophil cytoplasm antibodies (ANCA) Nephrol Dial Transplant. 1993;8:696–702. doi: 10.1093/ndt/8.8.696. [DOI] [PubMed] [Google Scholar]
- 39.Mellbye O, Mollnes T, Steen L. IgG subclass distribution and complement activation ability of autoantibodies to neutrophil cytoplasmic antigens (ANCA) Clin Immunol Immunopathol. 1994;70:32–39. doi: 10.1006/clin.1994.1007. [DOI] [PubMed] [Google Scholar]
- 40.Tosi M, Berger M. Functional differences between the 40 KDa and 50–70 KDa IgG Fc receptors on human neutrophils revealed by elastase treatment and antireceptor antibodies. J Immunol. 1988;141:2097–103. [PubMed] [Google Scholar]
- 41.Huizinga T, Dolman K, van der Linden N, et al. Phosphatidylinositol-linked FcRIII mediates exocytosis of neutrophil granule proteins, but does not mediate initiation of the respiratory burst. J Immunol. 1990;144:1432–7. [PubMed] [Google Scholar]
- 42.Salmon J, Edberg J, Kimberly R. Fcγ receptor III on human neutrophils. Allelic variants have functionally distinct capacities. J Clin Invest. 1990;85:1287–95. doi: 10.1172/JCI114566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kocher M, Edberg JC, Fleit HB, Kimberly RP. Antineutrophil cytoplasmic antibodies preferentially engage FcγRIIIb on human neutrophils. J Immunol. 1998;161:6909–14. [PubMed] [Google Scholar]
- 44.Fijen C, Berdius R, Kuijper E. Polymorphism of IgG Fc receptors in meningococcal disease. Ann Intern Med. 1993;119:636. doi: 10.7326/0003-4819-119-7_part_1-199310010-00026. [DOI] [PubMed] [Google Scholar]
- 45.Zhou M, Brown E. CR3 (Mac-1 aMb2 CD11b/CD18) and FcγRIII cooperate in generation of a neutrophil respiratory burst: requirement for FcγRII and tyrosine phosphorylation. J Cell Biol. 1994;125:1407–16. doi: 10.1083/jcb.125.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salmon J, Brogle N, Edberg J, Kimberley R. Fcγ receptor III induces actin polymerisation in human neutrophils and primes phagocytosis mediated Fcγ receptor II. J Immunol. 1991;146:997–1004. [PubMed] [Google Scholar]
- 47.Brennan P, Zigmond S, Schreiber A, Smith E, Southwick F. Binding of IgG containing immune complexes to human neutrophil FcγRII and FcγRIII induces actin polymerisation by a pertussis toxin-insensitive transduction pathway. J Immunol. 1991;146:997–1004. [PubMed] [Google Scholar]
- 48.Boros P, Odin J, Muryoi T, Masur S, Bona C, Unkeless J. IgM anti-Fc gamma R autoantibodies trigger neutrophil degranulation. J Exp Med. 1991;173:1473–82. doi: 10.1084/jem.173.6.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]