

Abstract
longitudinals-lacking (lola) was identified in Drosophila as a gene encoding several alternatively spliced transcription factors involved in axon guidance. Here we report that lola also plays a critical role in programmed cell death in the ovary. lola mutant germline clones show a high percentage of egg chambers with nurse cell nuclei persisting past stage 13, indicating a block in developmental nurse cell death. Mutants also show a disruption in the induced programmed cell death that occurs during mid-oogenesis in response to starvation. Further characterization revealed that lola germline clones exhibit abnormal nuclear organization which becomes increasingly severe with age. Chromatin appears diffuse and fails to condense properly or undergo DNA fragmentation in dying nurse cells. Masses of nuclear material accumulate in the ovaries of older flies containing lola germline clones. We propose that lola is necessary for complete chromatin condensation which occurs during programmed cell death in the ovary. Alleles differed in the strength of their phenotypes but interestingly, the severity of their ovarian phenotypes was independent of the strength of their neuronal phenotypes, suggesting a differential requirement for individual lola isoforms in the ovary and nervous system.
Keywords: Programmed cell death, Apoptosis, Oogenesis, Drosophila, lola, Germline clones, Chromatin condensation, Nurse cells
Introduction
In Drosophila, the process of oogenesis is divided into 14 stages based on well defined morphological characteristics (King, 1970; Spradling, 1993). A Drosophila ovary consists of 15–20 ovarioles, each containing several egg chambers at different stages of development. Each egg chamber is surrounded by somatically-derived follicle cells and contains 15 germline-derived nurse cells in addition to the oocyte. The nurse cells serve to provide the developing oocyte with essential proteins, organelles, and other cellular components.
Programmed cell death (PCD) is known to occur during early, mid-stage, and late-stage Drosophila oogenesis (reviewed in McCall, 2004). In response to nutrient deprivation, germline cyst cells may undergo PCD in the beginning of oogenesis in the germarium, or entire egg chambers may die around stage seven or eight, indicating that there are checkpoints at specific stages of oogenesis. During late Drosophila oogenesis, as a part of normal egg chamber development, the nurse cells undergo PCD resulting in a mature oocyte surrounded by the follicle cells that make the eggshell. Cytoskeletal changes occur as nurse cell PCD initiates, allowing for the transfer of cytoplasm from the nurse cells through intracellular bridges into the oocyte, a process commonly referred to as “dumping”. After the nurse cells transfer their mRNA, organelles, and proteins into the developing oocyte, the remaining nurse cell nuclei undergo chromatin condensation and DNA fragmentation (Cavaliere et al., 1998; Foley and Cooley, 1998; McCall and Steller, 1998). Eventually the nurse cell remnants are engulfed by the neighboring follicle cells (Cummings and King, 1970; Nezis et al., 2000).
Although these are well established characteristics of PCD in the ovary, it is currently unclear what role nurse cell DNA fragmentation plays, along with chromatin condensation, in the PCD process. Drosophila mutants lacking caspase-activated DNase (CAD) failed to undergo nucleosomal DNA fragmentation during oogenesis, although the ovaries did not accumulate DNA, and the nurse cells appeared to complete PCD (Mukae et al., 2002). Upon induction of apoptosis, lack of DNA fragmentation was also observed in human cell lines expressing a caspase-resistant form of CAD inhibitor (ICAD), which is normally cleaved by caspases to release CAD (McIlroy et al., 1999). However, nucleosomal DNA fragmentation was still present in mice expressing a caspase-resistant ICAD (McIlroy et al., 2000). Flies containing a mutation in the lysosomal acid DNase gene, dDNaseII, exhibited enhanced DNA fragmentation along with an accumulation of nurse cell DNA in the ovary. In addition, flies lacking CAD and DNaseII also showed DNA accumulation (Mukae et al., 2002). These results suggest that CAD and DNaseII may be involved in a two-step process mediating DNA fragmentation in the ovary (Mukae et al., 2002) similar to the three-step process proposed for C. elegans (Wu et al., 2000).
Previous studies have shown that many of the genes known to play essential roles in PCD during Drosophila embryogenesis and organogenesis are not necessary for PCD during oogenesis (Foley and Cooley, 1998; reviewed in Baum et al., 2005). This suggests that a different set of genes may be controlling this process in the ovary. Furthermore, the checkpoint PCD pathway active in mid-oogenesis appears to require different genes than the developmental PCD pathway active in later stages (reviewed in McCall, 2004). Over-expression of the inhibitor of apoptosis protein, Diap-1, has been shown to significantly block the mid-stage checkpoint cell death pathway, but has a milder effect on late-stage nurse cell death (Peterson et al., 2003; Mazzalupo and Cooley, 2006; J. Baum and KM, unpublished). Also, mutations in the effector caspase Dcp-1 prevent starvation induced PCD but not late-stage developmental PCD (Laundrie et al., 2003). In fact, relatively few genes involved in late stage developmental PCD have been identified thus far.
To identify genes necessary for proper nurse cell developmental PCD, we performed a germline clone (GLC) screen of chromosome 2R. One of the genes identified through this screen was longitudinals-lacking (lola). Lola was previously identified as a transcription factor involved in axon guidance (Giniger et al., 1994; Madden et al., 1999; Crowner et al., 2002; Goeke et al., 2003). Specifically, Lola has been shown to regulate the expression of axon guidance proteins Robo and Slit (Crowner et al., 2002). Furthermore, Lola has been shown to interact with chromosomal kinase JIL-1 (Zhang et al., 2003b), suggesting that it is also involved in chromatin structure modification and its roles in development could be more broad than initially thought. The lola gene spans over 61 kb, contains 32 alternatively spliced exons, and is known to encode at least 20 different isoforms (Goeke et al., 2003; Ohsako et al., 2003). Transcripts for all 20 isoforms contain common exons 5–8, which encode a BTB (Bric-a-brac, Tramtrack, Broad complex) domain involved in dimerization. lola additionally contains four alternatively spliced 5’ exons and 24 alternatively spliced 3’ exons. The 3’ variable exons encode several unique zinc finger motifs present in 17 of the 20 isoforms (Goeke et al., 2003; Horiuchi et al., 2003; Ohsako et al., 2003). lola has been shown to exhibit intragenic complementation through interallelic trans-splicing events, indicating that this is a very large and complex locus (Horiuchi et al., 2003).
Here we show that lola is also required for proper PCD in the ovary. Through GLC analysis we have determined that loss of lola results in a block in the developmental PCD which occurs late in oogenesis. Interestingly, lola mutants also show a delay in checkpoint PCD which occurs during the mid-stages, suggesting lola affects events common to both forms of PCD. Specifically we have observed a disruption in normal nurse cell nuclear organization, chromatin condensation, and DNA fragmentation that normally occur during PCD. We also show a differential requirement for individual lola splice forms in the ovary and in embryonic development of the nervous system. We hypothesize that Lola isoform K is specifically required for nuclear organization, chromatin condensation, and DNA fragmentation in the ovary during PCD.
Materials and methods
Drosophila strains
All fly lines were obtained from the Bloomington Stock Center unless otherwise noted. Seven deficiency lines and 35 lethal alleles were used for complementation analysis including btbk09901 and lola00642. lola5D2, lola4E4 (Giniger et al., 1994), lolaORC4, lolaORE50 (Goeke et al., 2003), and lolaORE120 (Crowner et al., 2002) were gifts from Ed Giniger. The BB127 enhancer trap line (Schüpbach and Wieschaus, 1991) was a gift from Trudi Schüpbach. yw67c23 flies served as a wild-type control. Nutrient deprivation experiments were carried out as described (Peterson et al., 2003). All crosses were carried out at 25°C on standard fly food unless otherwise noted.
Genetic manipulations
The screen from which lola629 was isolated was carried out in the following manner. Males from two different isogenic dp FRTG13/CyO recombinant fly lines were subjected to 35mM EMS in 10% sucrose overnight. These males were then crossed to y2 ras v RpII215shits/FM7, l ; Gla/CyO virgin females, resulting in only female progeny. In the F1 generation, single non-Gla females were crossed with y w hsflp BB127; FRTG13 ovoD/CyO males containing a heat-shock inducible flippase, the BB127 enhancer trap encoding nuclear β-galactosidase, and FRTG13 marked with the dominant female sterile mutation ovoD. On days 5 and 6 or as soon as third instar larvae were visible, progeny were heat shocked in a 37° water bath for 1 hour. y w hsflp BB127/FM7, l or +; dp FRTG13/ FRTG13 ovoD females were collected from the F2 generation and conditioned on wet yeast paste along with males. These potential GLC containing females were dissected after being conditioned 3–10 days. Siblings were used to make balanced stocks of each screen line that exhibited an oogenesis phenotype as determined through DAPI staining.
To generate GLCs of existing lola alleles, the FRT site from FRTG13 L/CyO flies was recombined onto lola stocks. Correct stocks were determined through eye color, loss of L, and failure to complement the original lola stock and other alleles.
Staining procedures
For antibody and DAPI staining, females were conditioned on wet yeast paste and ovaries were dissected, fixed, and stained as described (Verheyen and Cooley, 1994) except they were mounted in Vectashield with DAPI (Vector Labs, Burlingame, CA). For propidium iodide staining following the Cleaved Caspase-3 antibody staining procedure, fixed tissue was incubated with 600 μg/ml RNase in PBS for 2 hours and stained with 1 μg/ml propidium iodide (Molecular Probes, Eugene, OR) in PBS for 30 minutes in the dark, washed 2 x 15 minutes in PBS, and mounted in Vectashield (Vector Labs, Burlingame, CA). Cleaved Caspase-3 antibody (Cell Signaling Technology, Danvers, MA) was used at a 1:100 dilution. Secondary antibody goat-anti-rabbit-Cy3 (Jackson ImmunoResearch Labs, West Grove, PA) was used at a 1:200 dilution. Lamin Dm0 antibodies ADL101 and ADL84.12 (developed by Paul A. Fisher and obtained from Developmental Studies Hybridoma Bank, Iowa City, Iowa) were used at a 1:1 dilution. Secondary antibody goat-anti-mouse-Cy3 (Jackson ImmunoResearch Labs, West Grove, PA) and secondary antibody goat-anti-mouse Alexa Fluor 488 (Molecular Probes, Eugene, OR) were used at a 1:200 dilution. For rhodamine-phalloidin staining, tissue was fixed as described (Verheyen and Cooley, 1994), then rinsed with Drosophila Ringer’s solution 2 x 10 minutes. Samples were then incubated in the dark for 20 minutes with 50-100 μl rhodamine-phalloidin (Molecular Probes, Eugene, OR) made by diluting a 10 μl aliquot with 100 μl Ringer’s. Rhodamine-phalloidin was removed and samples were rinsed 2 x 1 minute with PBS and mounted in Vectashield with DAPI (Vector Labs, Burlingame, CA). Alternatively, for double staining with Lamin Dm0 ADL84.12 and rhodamine-phalloidin, tissue was rinsed 2 x 10 minutes in Drosophila Ringer’s solution following Lamin Dm0 antibody staining procedure. Tissues were then incubated with rhodamine-phalloidin as described above. The embryonic CNS was visualized with the BP102 antibody (developed by C. Goodman and obtained from Developmental Studies Hybridoma Bank, Iowa City, Iowa), used at a 1:3 dilution. For BP102 antibody staining, embryos from heterozygous adult flies were collected on egg plates overnight, fixed and stained as described (Patel, 1994). X-gal staining was carried out as described (McCall and Steller, 1998). TUNEL analysis was carried out as described (McCall and Peterson, 2004). Images were taken using an Olympus MagnaFire SP digital camera for samples viewed on an Olympus BX60 microscope, while confocal images were taken on an Olympus Fluoview confocal microscope.
Western Blotting
Extracts of total embryonic or ovarian protein were prepared using 40 14–17 hour old embryos or 4 pairs of ovaries for each genotype and separated by SDS-PAGE. Western analysis was performed by standard methods using anti-Lola antibody (Giniger et al., 1994) at a 1:350 dilution and visualized by enhanced chemiluminescence (ECL). Lamin Dm0 antibody ADL101 (developed by Paul A. Fisher and obtained from Developmental Studies Hybridoma Bank, Iowa City, Iowa) was used as a loading control at a 1:50 dilution.
DNA Sequencing
lola629 flies were balanced over CyO, Kr-GAL4 UAS-GFP. DNA was obtained from 14-17 hour old non-GFP embryos and amplified through PCR. Primer pairs used for amplification of lola exons 21 and 22 were 5’GAACTAAAACGAAAACGGAACC3’ and 5’GATGGGGATGTATGATTGTCG3’, 5’TCATCGGAATGCGGTAAGTC3’ and 5’TGATGGACCGTTTGCAGTTC3’, 5’CCATCCAGCATCAACATCAC3’ and 5’CTCACAATCCTCCCAACTTTC3’. PCR products were purified using QIAquick PCR purification kit (Qiagen, Valencia, CA) and sent to MWG-Biotech, Inc. (High Point, NC) for DNA sequencing. The background strain used for the screen was sequenced in a similar manner.
Results
Screen line 629 displays dumpless egg chambers, persisting nurse cell nuclei, and too many nurse cells
To identify genes involved in PCD in the Drosophila ovary, a genetic screen was undertaken in which GLCs were made of chromosome 2R after EMS mutagenesis (see Materials and methods). This homozygous mutant ovary tissue was then stained with DAPI and screened for abnormal PCD phenotypes such as egg chambers with altered numbers of nurse cells, excessive degeneration, or nurse cells surviving past stage 13 of oogenesis.
Screen line 629 exhibited a variety of interesting oogenesis defects, with 90% of the stage 14 egg chambers appearing abnormal. The major phenotypes observed were dumpless egg chambers or egg chambers with persisting nurse cell nuclei. Dumpless refers to stage 14 egg chambers in which the nurse cells have failed to transfer their cytoplasmic contents into the oocyte. In these egg chambers, the nurse cell nuclei are still intact and do not appear to be undergoing PCD (Fig. 1B). Persisting nurse cell nuclei refers to stage 14 egg chambers in which the nurse cells have transferred their cytoplasmic contents to the oocyte, but their nuclei have subsequently failed to undergo PCD including nuclear condensation, DNA fragmentation and/or engulfment. The only difference in appearance between these two categories of egg chambers is the presence or absence of cytoplasm. Additionally, some egg chambers were “cup-shaped,” mature egg chambers in which the chorion failed to extend around and seal the anterior end of the oocyte resulting in an open-ended chorion phenotype (Schüpbach and Wieschaus, 1991). These cup-shaped egg chambers also appear to have dorsal appendage material which has not elongated properly. In addition to these three major phenotypes, some early and mid-stage egg chambers in screen line 629 contain more than 15 nurse cells, a phenotype that could be due to altered PCD in the germarium (Smith et al., 2002), fusion of egg chambers, or too many cell divisions. The mutant GLCs also showed an excessive amount of degeneration in mid-oogenesis.
Fig. 1.

lola alleles vary in the severity of their GLC phenotypes. All egg chambers have been DAPI-stained. (A) Control, yw stage 14 egg chambers do not contain any remaining nurse cell nuclei. (B–H) Representative stage 14 egg chambers containing GLCs of the indicated lola alleles show that most cause moderate to severe defects during oogenesis including dumpless egg chambers. However, lola4E4 (C) looks wild-type.
Line 629 contains a lethal mutation mapping to the lola locus
Complementation analysis was performed with deficiencies spanning chromosome 2R, and line 629 was found to fail to complement Df(2R)stan1. After testing complementation with five smaller deletions mapping to the same region, we tested line 629 for complementation with several lethal alleles mapping to the appropriate region of 2R. Line 629 failed to complement two mutations including lola00642, and a P-element insertion line known as btbk09901 (bumper-to-bumper). Although btbk09901 had been found to complement other alleles of lola (Prokopenko et al., 2000), our data indicate that btbk09901 is an allele of lola. btbk09901 shows nervous system defects (Kania et al., 1995) similar to alleles of lola (Giniger et al., 1994; Madden et al., 1999; Crowner et al., 2002; Goeke et al., 2003; Horiuchi et al., 2003), and we have found that btbk09901 fails to complement several alleles of lola including lola00642, lola5D2, lolaORC4, lolaORE50, lola4E4, and lolaORE120 (data not shown). Based on these results, we conclude that 629 is an allele of lola.
lola is a large gene with 32 exons spanning over 61 kb, making it difficult to pinpoint the precise location of the mutation in line 629 (hereafter referred to as lola629). However, lola alleles are known to exhibit intragenic complementation through trans-splicing (Horiuchi et al., 2003), so we used this phenomenon to our advantage when mapping the mutation in lola629. Trans-splicing occurs when exons transcribed from one chromosome are spliced together with exons transcribed from the homologous chromosome to make a complete mRNA. As a result, mutations in different exons can complement one another because normal mRNA can still be made. Complementation analysis was performed using five additional lola alleles, several of which have demonstrated intragenic complementation (Horiuchi et al., 2003). Fig. 2A shows the results of this study. lola629 failed to complement all lola alleles that were tested with the exception of lola4E4 and lolaORE120. lola4E4, a P-element allele containing an insertion upstream of 5’ variable exon 3 (Giniger et al., 1994; Fig. 2B), is described as a strong allele (Giniger et al., 1994) and we found that it failed to complement other alleles in our study including lola5D2 and lola00642. lolaORE120 is a weaker EMS allele mapping to the constant region which includes exons 5–8 (Crowner et al., 2002; Fig. 2B).
Fig. 2.
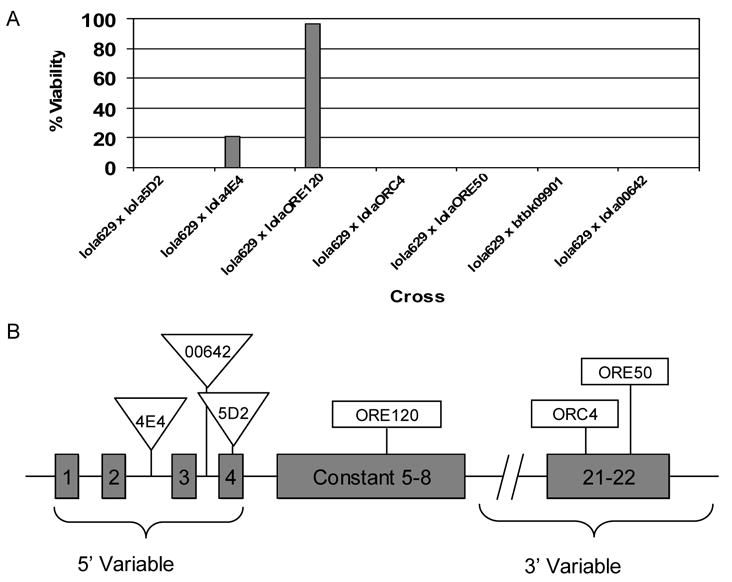
Intragenic complementation analysis. (A) Complementation results with different lethal alleles of lola helped narrow down the location of the lethal mutation in lola629. Offspring from the F1 generation of the indicated cross were counted. Percent viability is calculated as 100 x (observed number of straight winged flies/ expected number of straight winged flies). Expected number of straight winged flies = 1/2(observed number of curly winged flies) (Horiuchi et al., 2003). (B) Locations of P-element insertions in lola4E4, lola00642, and lola5D2, and EMS-induced mutations in lolaORE120, lolaORC4, and lolaORE50. Only relevant exons are depicted and drawing is not to scale (Horiuchi et al., 2003).
Importantly, lola629 failed to complement both lolaORC4 and lolaORE50 which harbor mutations mapping to the 3’ variable exon 22 included in isoform K (known as lola-PI, FlyBase) (Goeke et al., 2003). Horiuchi et al. (2003) previously determined that alleles with mutations mapping to the 3’ variable regions are likely to complement alleles with mutations mapping to 3’ variable exons corresponding to different isoforms. Furthermore, it was determined that mutations mapping to the 3’ variable region are likely to complement mutations mapping to the constant region of lola including lolaORE120 because of trans-splicing (Horiuchi et al., 2003). Because lola629 showed the same complementation pattern as lolaORC4 and lolaORE50, we predicted that the mutation in lola629 would also map to the 3’ variable region encoding isoform K which includes exons 21 and 22. Interestingly, lolaORC4 and lolaORE50 are known to have far more subtle neuronal defects than other alleles, and in particular, they lack the severe CNS phenotype that lola is named for (Goeke et al., 2003). lola629 was also found to lack this neuronal phenotype (Table 1), providing further support for our hypothesis that the lola mutation was in exons encoding isoform K.
Table 1.
Comparison of ovarian and neuronal phenotypes in lola alleles
| Relative phenotypic severity
|
||
|---|---|---|
| Genotype | Ovariana | Neuronal |
| lola629 | +++ | − (this study, data not shown)b |
| lola4E4 | − | +++ (Giniger et al., 1994) |
| lolaORE120 | +++ | + (Crowner et al., 2002) |
| lola5D2 | ++ | +++ (Giniger et al., 1994) |
| lola00642 | +++ | +++ (this study, data not shown) |
| lolaORC4 | +++ | +/− (Goeke et al., 2003) |
| lolaORE50 | NDd | +/− (Goeke et al., 2003) |
| btbk09901 | + | ++ (Kania et al., 1995) |
| FRTG13c | − | ND |
+++ indicates a strong mutant phenotype, ++ and + indicate moderate phenotypes, +/− indicates a weak phenotype, and – indicates no abnormal phenotype
All ovarian phenotypes are based on GLC analysis
This result is based on the presence of longitudinal axons in homozygous mutant embryos. Other possible neuronal phenotypes have not been examined.
examined for an ovarian phenotype as a control
Not determined
Upon sequencing exons 21 and 22 using DNA from lola629 homozygous embryos, a C to T mutation in exon 22 was found which was not present in the original strain used for the EMS mutagenesis screen. This mutation resulted in a glutamine being replaced with a stop codon at residue 635 of the K isoform of Lola. This isoform contains two zinc finger domains near the C-terminus (Goeke et al., 2003; Ohsako et al., 2003), and the early stop codon would terminate the protein over 250 residues upstream of these zinc finger domains, most likely disrupting the function of this protein. The finding that lola629 contains an isoform-specific mutation suggests that isoform K plays an important role during PCD in the ovary.
lola alleles differ in the severity of their ovarian phenotype
Because lolaORC4, lolaORE50, and lola629 each exhibit a weak neuronal phenotype (Table 1), and lola629 shows a very strong ovarian phenotype (Fig. 1B, Table 1), we wished to determine if other isoform K alleles show this strong ovarian phenotype as well. We also wanted to see if there was a relationship between the severity of neuronal phenotypes and severity of ovarian phenotypes among the lola alleles which would indicate isoform specific requirements in these two areas of development. Therefore, GLCs were made from six lola alleles in addition to lola629. Fig. 1 includes representative pictures of stage 14 egg chambers containing GLCs of these alleles along with a wild-type stage 14 egg chamber for comparison (Fig. 1A). The results indicate that most lola alleles do cause dumpless egg chambers and/or persisting nurse cell nuclei (Fig. 1B, and 1D–H). However, although lola4E4 shows severe neuronal phenotypes (Giniger et al., 1994), and our recombinant FRT lola4E4 line retained a strong CNS phenotype (data not shown), surprisingly we did not detect a significant phenotype in the ovary (Fig. 1C). Fig. 3 shows a quantitative analysis of the major phenotypes observed in these GLCs in comparison with wild-type GLCs (Fig. 3, FRTG13). Interestingly, the isoform K specific mutants lola629 and lolaORC4 have the strongest ovarian phenotype (Fig. 3). lolaORE120 is thought to be a weak allele and shows only a mild neuronal phenotype (Crowner et al., 2002). However, lolaORE120 GLCs also exhibited a significant ovary phenotype (Fig. 3, Table 1). Strong alleles thought to disrupt function of all Lola isoforms, with the exception of lola4E4, show both the severe neuronal and ovarian phenotypes. These alleles include lola5D2 and lola00642 (Fig. 3, Table 1). btbk09901 also exhibits a neuronal phenotype and was found to have a significant oogenesis phenotype (Fig. 3, Table 1). Taken together, these results suggest that isoform K is critical for oogenesis, but not for axon guidance during embryogenesis.
Fig. 3.
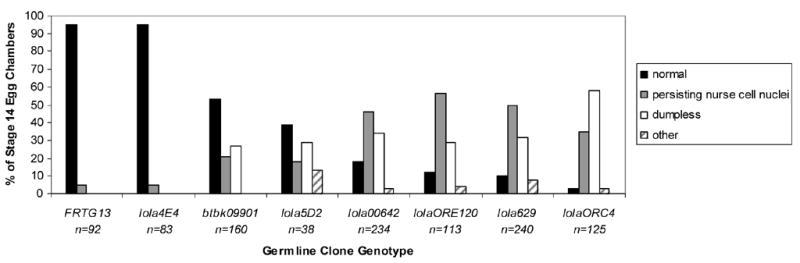
Quantification of ovarian phenotypes of different lola alleles in order of increasing severity. Stage 14 egg chambers were classified as one of four indicated categories. Those classified as other had dorsal appendage defects or were cup-shaped but did not contain persisting nurse cell nuclei and were not dumpless. FRTG13 germline clones were analyzed as a control. n= the total number of stage 14 germline clone egg chambers classified.
Lola isoform K is normally expressed during oogenesis and is missing in lolaORC4, lola629, and lolaORE50 mutants but not in lola4E4 mutants
To shed light on why isoform K specific alleles of lola exhibited strong ovarian phenotypes while lola4E4 mutant ovaries appeared wild-type, western analysis was done using protein extracted from homozygous embryos. It had previously been determined that lolaORC4 mutants express a truncated version of isoform K (Goeke et al., 2003). We wanted to see if this was also the case with lola629 mutants because these two alleles have very similar phenotypes. Using an antibody that recognizes a portion of Lola encoded by the common region (Giniger et al., 1994), we observed that lola629 and lolaORC4 homozygous embryos are each missing a large Lola isoform when compared with yw embryos. In addition, we observed a similar phenomenon in lolaORE50 embryos (Fig. 4A, arrowhead). This is consistent with each allele disrupting the expression of isoform K. It was also observed that like lolaORC4, lola629 mutants express a truncated version of this isoform seen as the appearance of a unique smaller band. Interestingly, these truncated proteins are expressed at a higher level than other Lola isoforms. The third isoform K specific allele, lolaORE50, also expressed a truncated version of isoform K, although it was not as highly expressed as those seen in lola629 and lolaORC4 embryos (Fig. 4A, arrows). These results suggest that lolaORC4 and lola629 have a strong ovarian phenotype due specifically to the lack of expression of isoform K.
Fig. 4.
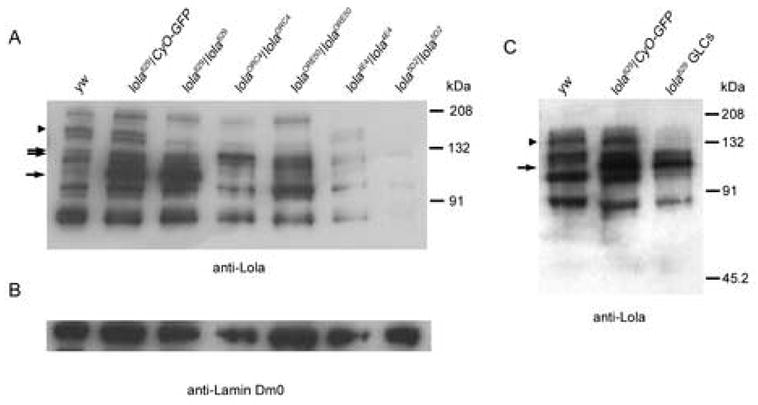
Lola isoform K expression is necessary for normal oogenesis. (A) Extracts from embryos of the genotypes indicated above were separated by SDS-PAGE and subjected to western analysis using an antibody that recognizes the constant region of Lola. The positions of standard protein molecular weight markers are indicated to the right. Compared with yw control embryos, those heterozygous and homozygous for lola629, as well as lolaORC4, and lolaORE50 embryos express truncated versions of isoform K (arrows). lola629, lolaORC4, lolaORE50, and lola5D2 homozygotes lack expression of the normal sized isoform K while lola4E4 retains expression (arrowhead). (B) The same membrane used for (A) was probed with an antibody for Lamin Dm0 to verify that comparable amounts of protein were added to each lane. (C) Protein extracts from ovaries of the genotypes indicated above were subjected to western analysis as in (A). Ovaries from lola629 heterozygotes and those containing GLCs of lola629 express a truncated version of isoform K (arrow), while only the GLCs lack expression of the normal sized isoform (arrowhead).
lola4E4 GLCs displayed no significant ovarian phenotype although lola4E4 has been previously considered a strong allele (Giniger et al., 1994). To determine if the lack of an ovarian phenotype was due to the retention of functional protein in lola4E4 mutants, protein from homozygous embryos was used in western analysis. It was determined that lola4E4 mutants still express many Lola isoforms, including isoform K, albeit at lower levels than in yw control embryos (Fig. 4A). In contrast, lola5D2, another strong lola allele (Giniger et al., 1994), had a strong ovarian phenotype. This allele also showed faint expression of some lola isoforms, however, importantly, there was little, if any, expression of isoform K (Fig. 4A). The presence of Lola isoform K in lola4E4, which unexpectedly did not show strong ovarian phenotypes, is further evidence that this isoform, in particular, is important during oogenesis.
To determine exactly which isoforms of Lola are expressed during oogenesis, western analysis was done again using the antibody against the common region of Lola that recognizes all isoforms (Giniger et al., 1994). Total protein extracts from yw and lola629 heterozygous ovaries as controls and ovaries containing GLCs of lola629 were probed (Fig. 4C). The results show that several isoforms are present in ovaries of control flies including isoform K. However, as was seen in homozygous embryos, this isoform is missing in lola629 GLCs (Fig. 4C, arrowhead) and a truncated version is apparent in both the GLCs and the heterozygous ovary extracts (Fig. 4C, arrow).
Mutations in lola block DNA fragmentation during mid-stage checkpoint and late-stage developmental programmed cell death
To determine specifically if the ovarian phenotypes observed in lola mutants were due to an absence, delay, or disruption of PCD, TUNEL (Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling) analysis was performed on GLCs. TUNEL analysis detects fragmented DNA, one of the hallmarks of PCD. For this, and all subsequent phenotypic analysis in the ovary, both lola629 and lolaORC4 GLCs were examined, due to the similar degree of abnormality and molecular nature of the alleles. When tested in parallel, no significant differences between the two alleles were observed. In addition, both heterozygous siblings of each allele and yw flies were used as “wild-type” controls (as noted in figures). No significant differences were observed between these controls.
To determine if lola played a role in the checkpoint cell death that occurs during mid-oogenesis, flies were starved by feeding them a 10% sucrose solution, and dying mid-stage egg chambers were analyzed by TUNEL and DAPI staining. The presence of DNA fragmentation, as depicted by dark-purple staining, was seen in the nurse cells of controls (Fig. 5A). Many degenerating mid-stage egg chambers were seen in lola mutant GLCs, even under well-fed conditions. However, although TUNEL-positive cells were seen in the surrounding ovoD tissue (Fig. 5B, arrows), most degenerating mid-stage lolaORC4 GLCs were not TUNEL-positive (Fig. 5C, arrows point to three individual degenerating egg chambers) indicating a block in DNA fragmentation during checkpoint PCD.
Fig. 5.
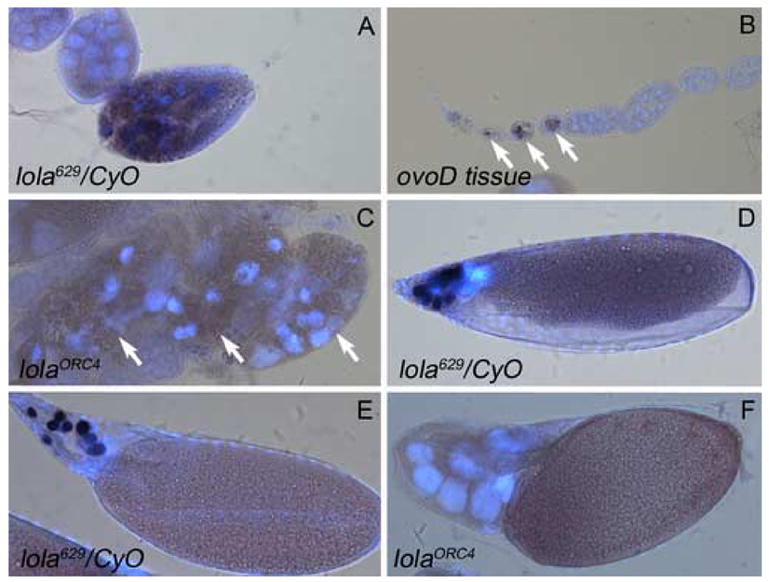
lola GLCs do not undergo DNA fragmentation during checkpoint or developmental PCD. TUNEL analysis (dark purple) and DAPI staining (blue) was performed on all egg chambers. (A) Heterozygous mid-stage egg chambers from nutrient deprived females undergo checkpoint PCD and show TUNEL positive nurse cell nuclei. (B) As an internal control, ovoD tissue, surrounding lolaORC4 GLCs, did contain TUNEL positive nuclei (arrows). (C) Mid-stage degenerating egg chambers containing lolaORC4 GLCs are not TUNEL positive. Arrows indicate three separate egg chambers. (D) Late stage 12 and (E) early stage 13 heterozygous control egg chambers display TUNEL positive nurse cells during developmental PCD. (F) lolaORC4 GLCs in a dumpless stage 14 egg chamber are not TUNEL positive.
During normal development, nurse cells become TUNEL-positive between late stage 12 and early stage 13, indicating the presence of fragmented DNA (Foley and Cooley, 1998; Fig. 5D and 5E). However, most dumpless egg chambers from lolaORC4 GLCs did not contain any TUNEL-positive nurse cells (Fig. 5F) indicating the absence of DNA fragmentation and incomplete developmental PCD. The same phenotypes were observed in lola629 GLCs (data not shown). These findings indicate that lola could affect a process common to both mid- and late oogenesis.
lola GLCs exhibit a disruption in the breakdown of nurse cell nuclear membranes
The first signs of nurse cell death in late oogenesis are convolution and permeability of the nuclear envelope (Cooley et al., 1992; Guild et al., 1997). This permeability can be visualized with a nuclear β-galactosidase (β-gal) marker which leaks out of nurse cell nuclei beginning during stage 10 of oogenesis (Cooley et al., ). Drosophila chickadee mutants are known to exhibit a dumpless phenotype because actin bundles fail to form and the nurse cells fail to transfer their cytoplasm through the ring canals into the oocyte (Cooley et al., 1992). Although this dumpless phenotype might suggest a disruption in PCD, the nurse cells do in fact undergo normal nuclear breakdown, as visualized with a nuclear ß-gal marker (Cooley et al., 1992).
To determine if the nurse cell nuclear envelope breaks down normally in dumpless egg chambers containing lola GLCs, the BB127 enhancer trap expressing nuclear β-gal in nurse cells was used (Schüpbach and Wieschaus, 1991). Proper breakdown of the nurse cell nuclear membranes would indicate a cytoskeleton disruption as seen in chickadee mutants, whereas failure to undergo nuclear breakdown would indicate a disruption in the process of PCD. In control, yw egg chambers, nurse cell nuclear membranes are intact through stage 10 (Fig. 6A). At stage 10B, the nuclear membranes break down, as indicated by the X-gal staining seen in the nurse cell cytoplasm (Fig. 6B). By stage 12, the nurse cells have transferred their cytoplasm to the oocyte, as indicated by the X-gal staining seen now in the oocyte (Fig. 6C). Finally at stage 14, all the nurse cell nuclei have undergone PCD, and the oocyte contains all of their cytoplasmic contents (Fig. 6D). In dumpless egg chambers containing lola629 GLCs, retention of X-gal staining inside nurse cell nuclei indicates that the nuclear membranes of the nurse cells fail to break down (Fig. 6E), suggesting that the dumpless phenotype is indeed due to a disruption in the process of PCD.
Fig. 6.
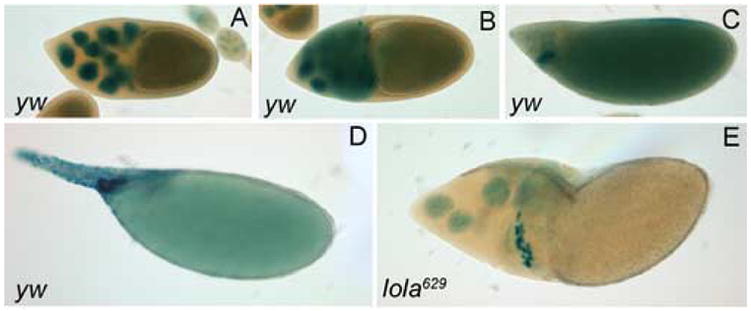
Nurse cell nuclear membranes do not break down in lola629 GLCs. (A–D) y w hsflp control and (E) lola629 GLC egg chambers contain the BB127 enhancer trap encoding nuclear β-galactosidase and are X-gal stained (blue). (A) A stage 10 yw egg chamber has intact nurse cell nuclear membranes. (B) By stage 10B, the nurse cell nuclear membranes break down and X-gal staining leaks into the cytoplasm. (C) A stage 12 yw egg chamber has undergone cytoplasmic transfer and X-gal staining is seen in the oocyte. (D) By stage 14, no nurse cells remain and all the X-gal staining is seen in the oocyte. (E) A dumpless egg chamber containing lola629 GLCs retains X-gal staining in the nurse cell nuclei.
Egg chambers containing lola GLCs show abnormal actin-cytoskeleton structure
During Drosophila oogenesis, cytoskeletal changes occur to facilitate rapid cytoplasm transfer from the nurse cells into the oocyte before the nurse cells complete developmental PCD. In healthy early and mid-stage egg chambers, subcortical actin surrounds each cell (Cooley et al., 1992; Nezis et al., 2006). In addition, actin filaments make up intercellular cytoplasmic bridges called ring canals that connect the nurse cells and oocyte. Actin bundles form in the cytoplasm of the nurse cells beginning at stage 10B and extend from the plasma membrane to the nuclear membrane. These actin bundles are thought to hold the nurse cell nuclei in place while the cytoplasm is transported through the ring canals (Cooley et al., 1992). Furthermore, these actin bundles are not normally seen during the mid-stage checkpoint PCD which does not involve any cytoplasmic transport (Nezis et al., 2000; Peterson et al., 2003).
To determine if the proper cytoskeletal changes occur in lola mutants during PCD, we used rhodamine-phalloidin staining to visualize actin. During the early stages of oogenesis, lola629 GLCs exhibit normal subcortical and ring canal actin (data not shown). Dying mid-stage egg chambers, stage 10B egg chambers, and dumpless egg chambers containing lola629 GLCs were also examined. Starvation induced PCD during mid-stages normally results in the formation of small clumps of actin (Fig. 7B, arrows) when nurse cell nuclei are condensed (Fig. 7A). However, degenerating egg chambers containing lola629 GLCs contain abnormal actin structures (Fig. 7D and 7F, white arrows). In heterozygous control degenerating mid-stage egg chambers, the nurse cells complete PCD before the surrounding follicle cells begin degenerating, as seen in wild-type (Fig. 7A and B, arrowheads). In contrast, most of the dying mid-stage egg chambers from lola629 GLCs (83%, n=127) had follicle cells which appeared to degenerate at the same or a faster rate than the nurse cells (Fig. 7D, arrowheads, compared with Fig. 7B, arrowhead). In addition, a portion of these egg chambers showed drastic differences in the degree of nurse cell nuclear condensation within one egg chamber, with uncondensed nuclei containing intact cellular membranes (Fig. 7E and 7F, yellow arrows). These results are consistent with mutations in lola causing a delay in the checkpoint PCD pathway as well as abnormal actin structure, and insufficient nuclear condensation. In lola GLC egg chambers, only the nurse cells and oocyte are homozygous for the mutation. The follicle cells, which are heterozygous, appear to die normally despite the lag in nurse cell death.
Fig. 7.
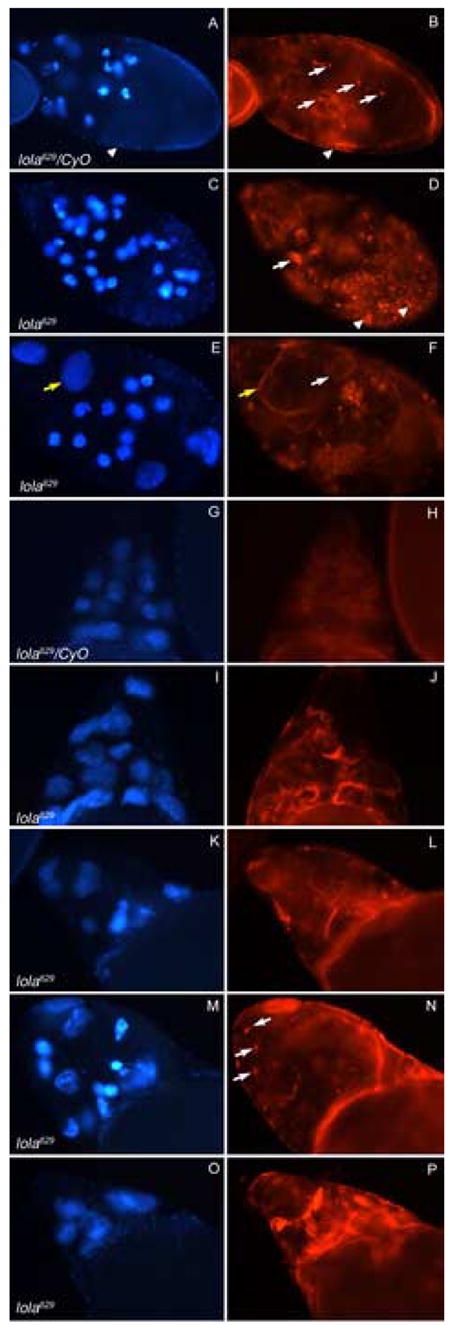
Abnormal actin structure is seen in degenerating lola629 GLC egg chambers. Panels depict DAPI staining (blue) on the left and rhodamine-phalloidin staining (red) on the right of the same egg chambers. All images were taken at the same magnification and rhodamine-phalloidin pictures were taken at the same exposure. (A–B) A heterozygous mid-stage egg chamber undergoing checkpoint PCD, induced through nutrient deprivation, contains condensed nurse cell nuclei, small actin clumps (B, arrows) and follicle cells with cellular membranes still intact (arrowheads). (C–F) lola629 GLC egg chambers undergoing checkpoint PCD, without starvation, contain abnormal actin structures (D and F, white arrows), degenerating follicle cells (D, arrowheads), and uncondensed nurse cell nuclei with intact cellular membranes (E and F, yellow arrows). (G–H) A stage 10B heterozygous egg chamber shows formation of organized actin bundles in all nurse cells. (I–J) A stage 10B egg chamber containing lola629 GLCs exhibits actin bundle formation that is disorganized and not uniform throughout the nurse cells. (K–P) Dumpless egg chambers containing lola629 GLCs continue to show disorganized actin structure. Some contain small actin clumps like those seen during mid-stage PCD (N, arrows).
During developmental PCD, heterozygous control stage 10B egg chambers exhibited normal actin bundle formation (Fig. 7G and 7H). However, stage 10B egg chambers containing lola629 GLCs showed disorganized actin bundles which were frequently much thicker than normal and not found uniformly throughout the egg chamber (Fig. 7I and 7J). Similar disorganization of the actin cytoskeleton was seen in wild-type egg chambers cultured in the presence of the caspase-3 inhibitor Z-DEVD-FMK, or treated with the cell death inducers etoposide or staurosporine (Nezis et al., 2006). Dumpless egg chambers continued to show disorganized actin structure (Fig. 7K–7P) including small actin clumps usually seen only during mid-stage checkpoint PCD (Fig. 7N, arrows). These results indicate that cytoskeletal changes normally occurring during developmental PCD to facilitate cytoplasmic transfer are disrupted in lola629 GLCs. Furthermore, the appearance of small actin clumps in dumpless egg chambers suggests the undying nurse cells may initiate the checkpoint PCD pathway as an alternative mechanism to die.
Mutations in lola cause variable levels of caspase activity during both checkpoint and developmental programmed cell death that are independent of chromatin condensation
To determine if lola alleles showed alterations in caspase activation in mid- or late oogenesis, egg chambers were labeled with Cleaved Caspase-3 antibody. This antibody has been found to detect robust caspase activity in egg chambers dying in mid-oogenesis (reviewed in Baum et al., 2005). Condensed nuclei and high levels of caspase activity were seen in heterozygous degenerating mid-stage egg chambers, induced to undergo PCD through nutrient deprivation of the adult females (Fig. 8A). Upon starvation of females containing lola629 or lolaORC4 GLCs, mid-stage egg chambers also showed high levels of caspase activation (Fig. 8B and 8C), however, it was not always seen throughout the entire egg chamber and sometimes localized to individual nurse cells (Fig. 8B). These results indicate that mutations in lola can cause a disruption in caspase activation and a delay in the checkpoint PCD pathway which is more severe in some nurse cells than in others, even within the same egg chamber.
Fig. 8.
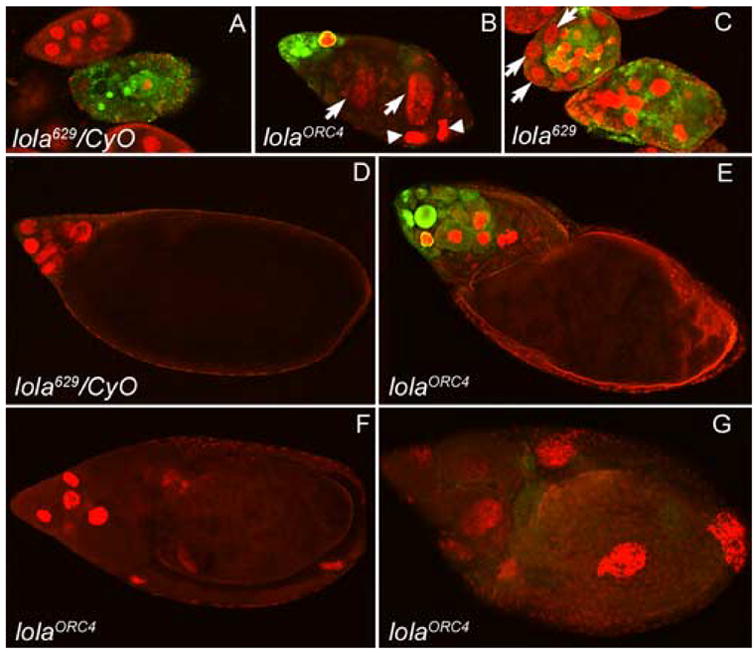
Variable levels of caspase activity are seen during checkpoint and developmental PCD in lola629 and lolaORC4 GLCs. Cleaved Caspase-3 antibody staining (green) depicts caspase activity and propidium iodide staining (red) shows nuclei. All images were taken at the same exposure and magnification. (A) A heterozygous mid-stage egg chamber undergoing checkpoint PCD, induced by nutrient deprivation, shows condensed nurse cell nuclei and high levels of caspase activity throughout the egg chamber. (B) Mid-stage egg chambers containing lolaORC4 or (C) lola629 GLCs, undergoing checkpoint PCD, also show high levels of caspase activity, but it is not uniformly found throughout the entire egg chamber. Nurse cells are not always condensed (B and C, arrows) and a lack of caspase activity is seen both with (B, arrowheads) and without (B and C, arrows) nuclear condensation. (D) A heterozygous stage 12 egg chamber undergoing developmental PCD does not show significant levels of caspase activity with this antibody. (E–G) Dumpless egg chambers containing lolaORC4 GLCs show either high levels of caspase activity (E), none (F), or very low levels (G). Lack of proper nuclear condensation can also be seen in some of these dumpless egg chambers (G).
Caspase activity in late oogenesis was found to be variable in lola629 and lolaORC4 GLCs. We did not detect any significant staining in control nurse cells undergoing developmental PCD in late oogenesis with this antibody (Fig. 8D). Similarly, some dumpless egg chambers from lola GLCs showed low or no caspase activity (Fig. 8F and 8G). However, others showed caspase activity at much higher levels than those seen during normal developmental PCD (Fig. 8E compared to 8D). These high levels of caspase activity may indicate the initiation of the checkpoint PCD pathway.
As noted before, in many lola GLC egg chambers undergoing PCD, normal condensation of individual nurse cell nuclei does not always occur (Fig. 8B and 8C, arrows, and 8G). Upon further investigation, we observed that both nurse cells displaying condensation (Fig. 8B, arrowheads, and 8F) and those without (Fig. 8B and 8C, arrows, and 8G) may lack caspase activity, suggesting that chromatin condensation can occur independently of caspase activation. Consistent with our findings, it was recently determined that incubation of developing wild-type egg chambers with the caspase-3 inhibitor Z-DEVD-FMK does not affect chromatin condensation although it does prevent DNA fragmentation and significantly disrupts development of the egg chambers (Nezis et al., 2006).
Chromatin structure defects in lola GLCs increase with age of the female
To determine how mutations in lola were specifically affecting oogenesis and chromatin condensation, a time-course experiment was performed to monitor the progression of lola629 GLC phenotypes. Flies were conditioned for 1 to 12 days before dissection. Fig. 9 shows representative pictures of ovaries from flies of different ages containing lola629 GLCs. In one day old flies, GLCs appeared normal (Fig. 9A). It is important to note that the lack of stage 14 egg chambers was expected due to the age of the flies. However, at two to three days (Fig. 9B–D), unusual nuclei with abnormal sizes and shapes are apparent in GLCs. By four days, masses of DNA were visible between GLC egg chambers (Fig. 9E, arrow). In seven to eight day old flies, the ovaries looked relatively messy and contained large masses of DNA (Fig. 9F, arrow). Upon closer examination of these masses, they appear to be the remnants of dying mid-stage egg chambers which have failed to undergo proper nurse cell chromatin condensation and engulfment. The follicle cells and cytoplasm are gone and all that is left is the nurse cell nuclei. When caspase activity was examined in these masses, it was clear that as the follicle cells die and the nurse cell cytoplasm disintegrates, the caspase activity leaves with it (Fig. 9G and 9H, arrows). It was also observed that these large masses contain very few TUNEL positive nuclei (data not shown), suggesting that proper and complete chromatin condensation is required for DNA fragmentation and is an essential part of PCD. These chromatin-related phenotypes are all seen in addition to the dumpless and persisting nurse cell nuclei phenotypes, however the severity of the latter phenotypes does not appear to be age-dependent.
Fig. 9.
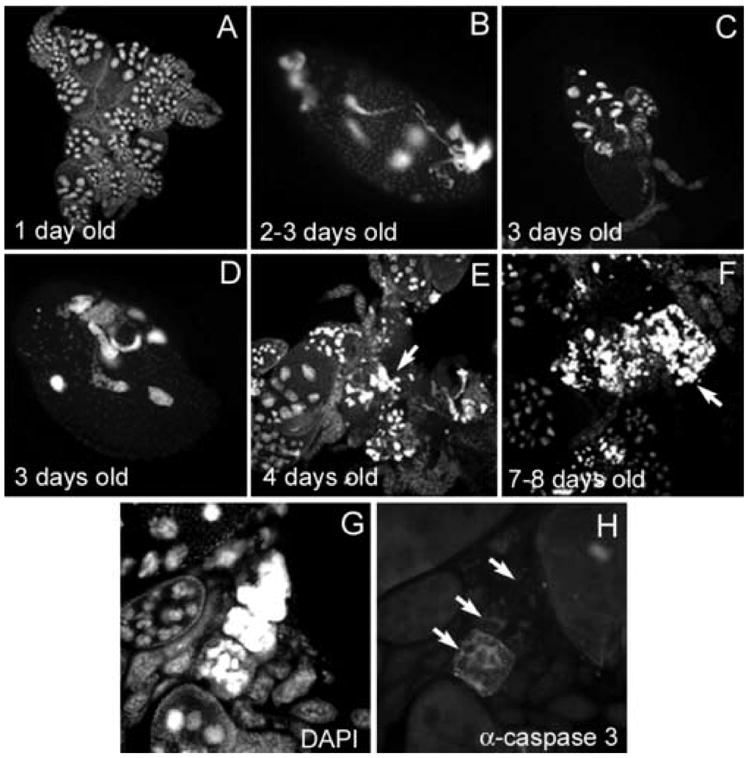
lola629 GLCs exhibit chromatin defects that increase with age of the female. Egg chambers in (A–G) were stained with DAPI and (H) was stained with Cleaved Caspase-3 antibody. (A) lola629 GLC tissue from a one day old female appears normal. (B–D) By two-three days, abnormally-shaped nuclei, along with nuclear condensation defects can be seen. (E) At four days, masses of nuclear material accumulate between egg chambers (arrow) and (F) by seven-eight days, these masses become larger (arrow). (G–H) A group of egg chambers stained with DAPI (G) and Cleaved Caspase-3 antibody (H) show loss of caspase activity after the follicle cells die and the nurse cell cytoplasm disperses (arrows). Remaining nuclear material in (G) may contribute to the masses in (E–F). (A), (C), and (E–F) were taken at the same magnification. (B), (D), and (G–H) were taken at higher magnification.
lola GLCs exhibit abnormal nuclear lamina morphology
A previous study showed that at least one Lola isoform interacts with chromosomal kinase JIL-1, implicating lola as being involved in chromatin structure regulation (Zhang et al., 2003b). Examination of JIL-1 mutants has revealed various defects in the ovaries including altered numbers of nurse cells, excessive degeneration, and defects in DNA amplification (Zhang et al., 2003a) as well as abnormal nuclear lamina morphology (Bao et al., 2005). JIL-1 is known to specifically regulate phosphorylation of serine 10 on histone H3 (Wang et al., 2001). Interestingly, we observed transient, stage specific labeling using an antibody recognizing this phosphorylation event, which coincided with the onset of developmental PCD in the nurse cells (data not shown). However, because the pattern of phosphorylation of serine 10 on histone H3 in nurse cells occurs specifically and transiently in later stage egg chambers, and because the majority of lola GLCs are abnormal by that stage, we were unable to compare levels of phosphohistone H3 (ser10) between lola GLCs and wild-type tissue. The involvement of lola in oogenesis stemming from a role it plays in chromatin structure regulation through interaction with JIL-1 seemed to be a strong possibility.
JIL-1 has also been found to interact with Lamin Dm0, and two distinct phenotypes observed in JIL-1 mutant ovaries were the mislocalization of nuclear lamins and an abnormal protrusion of the lamina through the ring canals near the oocyte (Bao et al., 2005). Furthermore, the abnormal nuclear lamina morphology in JIL-1 mutants was only observed in the nurse cells and not in any embryonic or larval tissues examined (Bao et al., 2005), suggesting that this interaction between JIL-1 and Lamin Dm0 is of particular importance in the ovary. To determine if the phenotypes observed in lola GLCs stem from an interaction between Lola and JIL-1, we performed lamin Dm0 antibody staining on both lola629 and lolaORC4 GLCs. Similar to JIL-1 mutant ovaries, we observed protrusions of nuclear lamina through ring canals in lola GLCs when egg chambers were double-labeled with lamin antibody (green) and phalloidin (red) (Fig. 10B). This phenotype was observed in 18% of mid-stage egg chambers that otherwise appeared normal (n=196), and these protrusions were never observed in heterozygous control egg chambers (Fig. 10A).
Fig. 10.
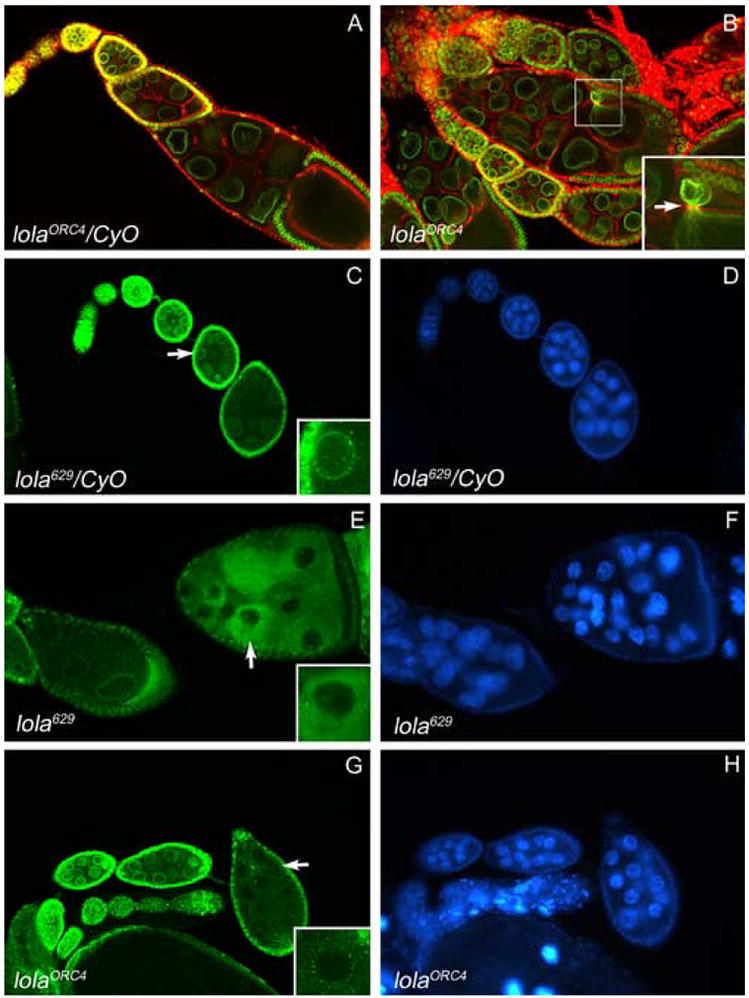
lola629 and lolaORC4 GLCs exhibit abnormal nuclear lamina morphology. Egg chambers in (A–B) were double-labeled with lamin Dm0 ADL84.12 antibody (green) and rhodamine-phalloidin (red). (A) Heterozygous control egg chambers exhibit normal nuclear lamina morphology. (B) A lolaORC4 GLC egg chamber exhibits a protrusion of the nurse cell nuclear lamina through the ring canal (arrow, higher magnification inset). Egg chambers in (C–H) were stained with lamin Dm0 ADL101 antibody (green, C, E, and G) and DAPI to visualize DNA (blue, D, F, and H). (C) Nuclear lamin Dm0 surrounds the nuclei in heterozygous control egg chambers (higher magnification inset of nucleus indicated by arrow). (E and G) lola629 and lolaORC4 GLCs exhibit mislocalization of lamin Dm0 to the cytoplasm ((higher magnification insets of nuclei indicated by arrows). (F and H) No abnormal chromatin morphology is seen in the egg chambers with mislocalized lamin Dm0 (compare with neighboring egg chambers and heterozygous controls in D).
We also observed an abnormal cytoplasmic localization of nuclear lamins in lola GLCs (Fig. 10E and 10G, arrows) when compared to heterozygous control egg chambers (Fig. 10C). This was seen in 22% of mid-stage egg chambers that otherwise appeared normal (n=156). Interestingly, the abnormal chromatin morphology frequently observed in lola GLCs was not present in the GLC egg chambers exhibiting this mislocalization of nuclear lamins (visualized by DAPI, Fig. 10F and 10H). Additionally, this phenotype was typically seen in early stages of oogenesis, suggesting that abnormal Lamin Dm0 distribution may precede chromatin condensation defects in lola GLCs. Furthermore, the observation of nuclear lamina morphology defects in lola GLCs similar to those seen in JIL-1 mutant ovaries suggests that Lola and JIL-1 may interact in the ovary.
Discussion
The results of this study indicate that the axon guidance gene lola is involved in PCD during both mid-oogenesis and late-oogenesis. These two forms of PCD have distinct characteristics (reviewed in McCall, 2004). The mid-oogenesis checkpoint PCD is inducible through starvation, and the entire egg chamber dies, including the nurse cells, oocyte, and follicle cells. On the other hand, during late-oogenesis, only the nurse cells undergo PCD as a natural part of the development of the egg chamber. In addition, these two forms of cell death have been shown to require different sets of genes. Despite these major differences, each form of PCD involves chromatin condensation, DNA fragmentation, and eventual engulfment of the nurse cell nuclear remnants by the surrounding follicle cells (reviewed in McCall, 2004).
It appears that lola is required for the chromatin condensation that occurs during the checkpoint PCD in mid-oogenesis. Without lola, egg chambers that have initiated the mid-stage checkpoint PCD do not undergo proper nuclear condensation, exhibiting either de-condensation, a complete lack of condensation, or an arrest at the condensed stage with no progression towards fragmentation or engulfment of the dying nurse cells. Although the presence of caspase activity and some chromatin condensation indicates the checkpoint PCD pathway has been initiated, it is incomplete. As a result, we see the degeneration of the surrounding follicle cells prior to the disappearance of the nurse cells and the accumulation of chromatin masses.
In late oogenesis, normal development leads to PCD of the nurse cells. However in lola mutants this pathway is not triggered normally. Actin bundles form improperly, cytoplasmic dumping fails to occur, and nurse cell nuclear membranes do not break down. Nuclei usually do not condense normally, similar to the condensation defects seen in mid-stage PCD, and consequently, the nurse cells do not die. Consistent with a block to this pathway, we see almost a complete absence of TUNEL-positive nuclei in these dumpless egg chambers. However, some dumpless egg chambers show high levels of caspase activation comparable to mid-stage degenerating egg chambers. We hypothesize that because caspases are activated to the same levels as during mid-stage checkpoint PCD, the dumpless egg chambers which show high levels of caspase activity may be undergoing this checkpoint PCD rather than the true developmental PCD. But without lola, there is a block or delay in the pathway once it is initiated, and although nurse cells sometimes appear to undergo chromatin condensation, the vast majority do not display any subsequent DNA fragmentation. This is further supported by the observance of small actin clump formation in some dumpless egg chambers which resembles mid-stage dying egg chambers.
Examination of the GLC phenotypes of several different alleles of lola revealed that mutations in the 5’ variable region, the constant region, and the 3’ variable region, specifically exon 22, can each cause severe defects in PCD during oogenesis. The severe ovarian phenotypes of both lola629 and lolaORC4 suggest that Lola isoform K plays a very important role during oogenesis and a relatively minor role during the development of the Drosophila nervous system. Both lola629 and lolaORC4 contain early stop codons located at residues 635 and 771 respectively, and both are located significantly before the zinc finger domains in isoform K (this study; Goeke et al., 2003; Horiuchi et al., 2003). The lolaORE50 allele contains a missense mutation at residue 745 (Goeke et al., 2003) and a four bp deletion causing a frameshift beginning at residue 777 (Goeke et al., 2003; Horiuchi et al., 2003), eventually ending in an early stop at residue 870. We were unable to generate GLCs of lolaORE50, however, it is likely that this allele would also show a major disruption during oogenesis.
In this study, intragenic complementation became a useful mapping strategy. In and of itself, the pattern of complementation displayed by lola and the evolution of its use of trans-splicing is decidedly complex and worthy of much more investigation. Modifier of mdg4 or mod(mdg4) is the only other Drosophila gene besides lola currently known to exhibit the phenomenon of interallelic complementation through trans-splicing (Dorn et al., 2001; Mongelard et al., 2002). Through this trans-splicing mechanism, mod(mdg4) has the ability, like lola, to produce many different gene products. In addition, mod(mdg4) has been shown to be involved in many different cellular processes including nerve pathfinding, position effect variegation, chromatin boundary establishment, and meiotic chromosome pairing (Dorn and Krauss, 2003). It is also interesting to note that Doom, a specific isoform of mod(mdg4), was identified in a screen for cellular factors involved in PCD (Harvey et al., 1997). Lola isoforms carry out a similarly wide variety of cellular functions. lola is thought to encode transcription factors implicated as playing important roles in both the central and peripheral nervous systems (Giniger et al., 1994; Madden et al., 1999; Crowner et al., 2002), chromatin structure (Zhang et al., 2003b), and oogenesis (this study; E. Giniger, personal communication). We have determined a requirement for lola in the germline which does not seem to be directly related to its requirements during embryonic development of the nervous system. These findings highlight how similar lola is to mod(mdg4) in carrying out a variety of functions.
Both lola and mod(mdg4) contain an N-terminal BTB domain (Dorn et al., 1993; Giniger et al., 1994). This domain is found in the constant region of lola and is contained in all isoforms (Goeke et al., 2003). Work in both C. elegans and S. pombe revealed that BTB domain containing proteins can interact with the ubiquitin ligase Cullin-3 (CUL-3) to target protein substrates for degradation (Furukawa et al., 2003; Geyer et al., 2003; Pintard et al., 2003; Xu et al., 2003). By directly binding to CUL-3 through their BTB domains, these proteins are thought to serve as adaptor molecules that recruit specific substrates to CUL-3. With substrate specificity being mediated by other domains found in these adaptors, a wide variety of substrates can be sequestered by this one ubiquitin ligase. If Lola interacts with dCul-3, this might explain how Lola isoforms can carry out such a diverse group of cellular functions. In addition to the BTB domain contained in every isoform, 17 Lola isoforms contain unique zinc finger domains (Goeke et al., 2003). One can hypothesize that each Lola isoform may be recruiting a different set of substrates to dCul-3 through these unique exons and thus targeting them for degradation. Recently guftagu (gft) was identified as the gene encoding Drosophila Cullin-3 (dCul-3) (Mistry et al., 2004). It was determined that high levels of dCul-3 are present in 0–1 hour old embryos suggesting a maternal contribution. Interestingly, flies containing GLCs of gft failed to lay most eggs and the ones that were laid appeared small and had dorsal appendage defects (Mistry et al., 2004). Observation of gft GLCs revealed a severe dumpless phenotype, however none of the chromatin defects seen in lola GLCs were present (data not shown). This suggests that Lola is not interacting with dCul-3 in the ovary.
We observed abnormal nuclear lamina morphology in isoform K specific lola GLCs. The abnormal nuclear lamina phenotype was apparent during early and mid-oogenesis and seemed to precede the other abnormalities described. Previous co-immunoprecipitation experiments have shown that at least one Lola isoform (known as lola-PE and lola-PD, FlyBase) can form a complex with chromosomal kinase JIL-1 (Zhang et al., 2003b). A genetic interaction between lola and JIL-1 has also been demonstrated using lola00642, an allele which is thought to knock out several isoforms. lola00642 acted as a dominant suppressor of JIL-1EP(3)3657 recessive semi-lethality, suggesting an antagonistic interaction (Zhang et al., 2003b). Further studies have shown that JIL-1 interacts with Lamin Dm0 in nurse cells (Bao et al., 2005). It has been suggested that the interaction between Lola and JIL-1 may be highly complex and dependent on the specific isoform of Lola and the developmental context (Zhang et al., 2003b). The same defects in the nuclear lamina that we saw in lola GLCs have been described in JIL-1 mutant ovaries, albeit with somewhat different penetrance. Specifically, protrusions of the nuclear lamina through the ring canal were observed in 42.8% of JIL-1 egg chambers, while mislocalized lamins were observed in 5.2% of JIL-1 egg chambers (Bao et al., 2005). However, overall JIL-1 mutant ovaries displayed nuclear lamin defects in 48% of egg chambers while lola GLCs displayed nuclear lamin defects in 40% of egg chambers, indicating that a positive interaction between Lola isoform K and JIL-1 may exist in the ovary. From this study, we cannot conclude that this is a direct interaction, but the involvement of lola in chromatin structure regulation during oogenesis stemming from an interaction with JIL-1 and consequently Lamin Dm0 remains a possibility.
Our studies suggest that lola isoform K is acting in a pathway leading to the regulation of chromatin condensation and completion of PCD during oogenesis. Analysis of other isoform specific lola mutants will help to determine which isoforms are needed for proper PCD during oogenesis and which are not. In addition, studies aimed at identifying targets of the different isoforms will help determine the molecular pathways in which Lola is acting during oogenesis.
Acknowledgments
We thank Ed Giniger for fly strains and the Lola antibody, and Trudi Schüpbach for fly strains. We are very grateful to the Bloomington Stock Center for providing us with the 2R deficiency kit and numerous other stocks. We thank members of our lab and Susan Tsunoda for helpful discussion and comments on the manuscript. This work was supported by National Institutes of Health grant R01 GM60574 to K.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bao X, Zhang W, Krencik R, Deng H, Wang Y, Girton J, Johansen J, Johansen KM. The JIL-1 kinase interacts with lamin Dm0 and regulates nuclear lamina morphology of Drosophila nurse cells. J Cell Sci. 2005;118:5079–87. doi: 10.1242/jcs.02611. [DOI] [PubMed] [Google Scholar]
- Baum JS, St George JP, McCall K. Programmed cell death in the germline. Semin Cell Dev Biol. 2005;16:245–59. doi: 10.1016/j.semcdb.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Cavaliere V, Taddei C, Gargiulo G. Apoptosis of nurse cells at the late stages of oogenesis. Dev Genes Evol. 1998;208:106–112. doi: 10.1007/s004270050160. [DOI] [PubMed] [Google Scholar]
- Cooley L, Verheyen E, Ayers K. chickadee encodes a profilin required for intercellular cytoplasm transport during Drosophila oogenesis. Cell. 1992;69:173–184. doi: 10.1016/0092-8674(92)90128-y. [DOI] [PubMed] [Google Scholar]
- Crowner D, Madden K, Goeke S, Giniger E. Lola regulates midline crossing of CNS axons in Drosophila. Development. 2002;129:1317–25. doi: 10.1242/dev.129.6.1317. [DOI] [PubMed] [Google Scholar]
- Cummings MR, King RC. Ultrastructural changes in nurse and follicle cells during late stages of oogenesis in Drosophila melanogaster. Z Zellforsch Mikrosk Anat. 1970;110:1–8. doi: 10.1007/BF00343981. [DOI] [PubMed] [Google Scholar]
- Dorn R, Krauss V. The modifier of mdg4 locus in Drosophila: functional complexity is resolved by trans splicing. Genetica. 2003;117:165–77. doi: 10.1023/a:1022983810016. [DOI] [PubMed] [Google Scholar]
- Dorn R, Krauss V, Reuter G, Saumweber H. The enhancer of position-effect variegation of Drosophila, E(var)3-93D, codes for a chromatin protein containing a conserved domain common to several transcriptional regulators. Proc Natl Acad Sci U S A. 1993;90:11376–11380. doi: 10.1073/pnas.90.23.11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn R, Reuter G, Loewendorf A. Transgene analysis proves mRNA trans-splicing at the complex mod(mdg4) locus in Drosophila. Proc Natl Acad Sci U S A. 2001;98:9724–9. doi: 10.1073/pnas.151268698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley K, Cooley L. Apoptosis in late stage Drosophila nurse cells does not require genes within the H99 deficiency. Development. 1998;125:1075–82. doi: 10.1242/dev.125.6.1075. [DOI] [PubMed] [Google Scholar]
- Furukawa M, He YJ, Borchers C, Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol. 2003;5:1001–7. doi: 10.1038/ncb1056. [DOI] [PubMed] [Google Scholar]
- Geyer R, Wee S, Anderson S, Yates J, Wolf DA. BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol Cell. 2003;12:783–90. doi: 10.1016/s1097-2765(03)00341-1. [DOI] [PubMed] [Google Scholar]
- Giniger E, Tietje K, Jan LY, Jan YN. lola encodes a putative transcription factor required for axon growth and guidance in Drosophila. Development. 1994;120:1385–98. doi: 10.1242/dev.120.6.1385. [DOI] [PubMed] [Google Scholar]
- Goeke S, Greene EA, Grant PK, Gates MA, Crowner D, Aigaki T, Giniger E. Alternative splicing of lola generates 19 transcription factors controlling axon guidance in Drosophila. Nat Neurosci. 2003;6:917–24. doi: 10.1038/nn1105. [DOI] [PubMed] [Google Scholar]
- Guild GM, connelly PS, Shaw MK, Tilney LG. Actin filament cables in Drosophila nurse cells are composed of modules that slide passively past one naother during dumping. J Cell Biol. 1997;138:783–797. doi: 10.1083/jcb.138.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey AJ, Bidwai AP, Miller LK. Doom, a product of the Drosophila mod(mdg4) gene, induces apoptosis and binds to baculovirus inhibitor-of-apoptosis proteins. Mol Cell Biol. 1997;17:2835–43. doi: 10.1128/mcb.17.5.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi T, Giniger E, Aigaki T. Alternative trans-splicing of constant and variable exons of a Drosophila axon guidance gene, lola. Genes Dev. 2003;17:2496–501. doi: 10.1101/gad.1137303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kania A, Salzberg A, Bhat M, D'Evelyn D, He Y, Kiss I, Bellen HJ. P-element mutations affecting embryonic peripheral nervous system development in Drosophila melanogaster. Genetics. 1995;139:1663–78. doi: 10.1093/genetics/139.4.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RC. Ovarian Development in Drosophila melanogaster. Academic Press; New York: 1970. [Google Scholar]
- Laundrie B, Peterson JS, Baum JS, Chang JC, Fileppo D, Thompson S, McCall K. Germline cell death is inhibited by P element insertions in the dcp-1/pita nested gene pair in Drosophila. Genetics. 2003;165:1881–1888. doi: 10.1093/genetics/165.4.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden K, Crowner D, Giniger E. LOLA has the properties of a master regulator of axon-target interaction for SNb motor axons of Drosophila. Dev Biol. 1999;213:301–13. doi: 10.1006/dbio.1999.9399. [DOI] [PubMed] [Google Scholar]
- Mazzalupo S, Cooley L. Illuminating the role of caspases during Drosophila oogenesis. Cell Death Differ. 2006 doi: 10.1038/sj.cdd.4401892. [DOI] [PubMed] [Google Scholar]
- McCall K. Eggs over easy: cell death in the Drosophila ovary. Dev Biol. 2004;274:3–14. doi: 10.1016/j.ydbio.2004.07.017. [DOI] [PubMed] [Google Scholar]
- McCall K, Peterson J. Detection of apoptosis in Drosophila. In: Brady H, editor. Apoptosis: Methods and Protocols. Vol. 282. The Humana Press, Inc; Totowa, NJ: 2004. pp. 191–206. [DOI] [PubMed] [Google Scholar]
- McCall K, Steller H. Requirement for DCP-1 caspase during Drosophila oogenesis. Science. 1998;279:230–234. doi: 10.1126/science.279.5348.230. [DOI] [PubMed] [Google Scholar]
- McIlroy D, Sakahira H, Talanian RV, Nagata S. Involvement of caspase 3-activated DNase in internucleosomal DNA cleavage induced by diverse apoptotic stimuli. Oncogene. 1999;18:4401–8. doi: 10.1038/sj.onc.1202868. [DOI] [PubMed] [Google Scholar]
- McIlroy D, Tanaka M, Sakahira H, Fukuyama H, Suzuki M, Yamamura K, Ohsawa Y, Uchiyama Y, Nagata S. An auxiliary mode of apoptotic DNA fragmentation provided by phagocytes. Genes Dev. 2000;14:549–58. [PMC free article] [PubMed] [Google Scholar]
- Mistry H, Wilson BA, Roberts IJ, O'Kane CJ, Skeath JB. Cullin-3 regulates pattern formation, external sensory organ development and cell survival during Drosophila development. Mech Dev. 2004;121:1495–507. doi: 10.1016/j.mod.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Mongelard F, Labrador M, Baxter EM, Gerasimova TI, Corces VG. Trans-splicing as a novel mechanism to explain interallelic complementation in Drosophila. Genetics. 2002;160:1481–7. doi: 10.1093/genetics/160.4.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukae N, Yokoyama H, Yokokura T, Sakoyama Y, Nagata S. Activation of the innate immunity in Drosophila by endogenous chromosomal DNA that escaped apoptotic degradation. Genes Dev. 2002;16:2662–71. doi: 10.1101/gad.1022802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezis IP, Stravopodis DJ, Margaritis LH, Papassideri IS. Chromatin condensation of ovarian nurse and follicle cells is regulated independently from DNA fragmentation during Drosophila late oogenesis. Differentiation. 2006;74:293–304. doi: 10.1111/j.1432-0436.2006.00076.x. [DOI] [PubMed] [Google Scholar]
- Nezis IP, Stravopodis DJ, Papassideri I, Robert-Nicous M, Margaritis LH. Stage-specific apoptotic patterns during Drosophila oogenesis. Eur J Cell Biol. 2000;79:610–620. doi: 10.1078/0171-9335-00088. [DOI] [PubMed] [Google Scholar]
- Ohsako T, Horiuchi T, Matsuo T, Komaya S, Aigaki T. Drosophila lola encodes a family of BTB-transcription regulators with highly variable C-terminal domains containing zinc finger motifs. Gene. 2003;311:59–69. doi: 10.1016/s0378-1119(03)00554-7. [DOI] [PubMed] [Google Scholar]
- Patel NH. Imaging Neuronal Subsets and Other Cell Types in Whole-Mount Drosophila Embryos and Larvae Using Antibody Probes. In: Goldstein LS, Fyrberg EA, editors. Methods in Cell Biology. Vol. 44. Academic Press, Inc.; San Diego, CA: 1994. pp. 445–487. [DOI] [PubMed] [Google Scholar]
- Peterson JS, Barkett M, McCall K. Stage-specific regulation of caspase activity in Drosophila oogenesis. Dev Biol. 2003;260:113–123. doi: 10.1016/s0012-1606(03)00240-9. [DOI] [PubMed] [Google Scholar]
- Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B, Peter M. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003;425:311–6. doi: 10.1038/nature01959. [DOI] [PubMed] [Google Scholar]
- Prokopenko SN, He Y, Lu Y, Bellen HJ. Mutations affecting the development of the peripheral nervous system in Drosophila: a molecular screen for novel proteins. Genetics. 2000;156:1691–715. doi: 10.1093/genetics/156.4.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüpbach T, Wieschaus E. Female sterile mutations on the second chromosome of Drosophila melanogaster. II. Mutations blocking oogenesis or altering egg morphology. Genetics. 1991;129:1119–36. doi: 10.1093/genetics/129.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JE, 3rd, Cummings CA, Cronmiller C. Daughterless coordinates somatic cell proliferation, differentiation and germline cyst survival during follicle formation in Drosophila. Development. 2002;129:3255–67. doi: 10.1242/dev.129.13.3255. [DOI] [PubMed] [Google Scholar]
- Spradling AC. Developmental genetics of oogenesis. In: Bate M, Martinez Arias A, editors. The Development of Drosophila melanogaster. I. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1993. pp. 1–70. [Google Scholar]
- Verheyen E, Cooley L. Looking at oogenesis. In: Goldstein LSB, Fyrberg EA, editors. Methods in Cell Biology. Vol. 44. Academic Press; New York: 1994. pp. 545–561. [PubMed] [Google Scholar]
- Wang Y, Zhang W, Jin Y, Johansen J, Johansen KM. The JIL-1 tandem kinase mediates histone H3 phosphorylation and is required for maintenance of chromatin structure in Drosophila. Cell. 2001;105:433–43. doi: 10.1016/s0092-8674(01)00325-7. [DOI] [PubMed] [Google Scholar]
- Wu YC, Stanfield GM, Horvitz HR. NUC-1, a caenorhabditis elegans DNase II homolog, functions in an intermediate step of DNA degradation during apoptosis. Genes Dev. 2000;14:536–48. [PMC free article] [PubMed] [Google Scholar]
- Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–21. doi: 10.1038/nature01985. [DOI] [PubMed] [Google Scholar]
- Zhang W, Jin Y, Ji Y, Girton J, Johansen J, Johansen KM. Genetic and phenotypic analysis of alleles of the Drosophila chromosomal JIL-1 kinase reveals a functional requirement at multiple developmental stages. Genetics. 2003a;165:1341–54. doi: 10.1093/genetics/165.3.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang Y, Long J, Girton J, Johansen J, Johansen KM. A developmentally regulated splice variant from the complex lola locus encoding multiple different zinc finger domain proteins interacts with the chromosomal kinase JIL-1. J Biol Chem. 2003b;278:11696–704. doi: 10.1074/jbc.M213269200. [DOI] [PubMed] [Google Scholar]