

Abstract
Complement component C6 is a part of the membrane attack complex that forms a pore-like structure in cell membranes following complement activation. Deficiency of terminal complement components including C6 predisposes individuals to infection with Neisseriae. Using polymerase chain reaction/single-strand conformation polymorphism analysis followed by DNA sequencing, we screened genomic DNA from 200 randomly chosen blacks and an equal number from whites for three loss-of-function C6 mutations. Ten blacks and two whites were found to be heterozygous for one of the mutations. Two of the mutations, 1195delC and 1936delG, were found exclusively in black individuals. A third previously undescribed mutation, 878delA, was found at equal frequency among the two groups. The difference between the two groups was significant (P = 0.027), indicating that C6 deficiency due to these three mutations is more common among blacks than whites in the local area, principally Jefferson County, Alabama. In addition, three previously undescribed point mutations, two of which result in amino acid substitutions, were identified within exon 6. A review of the county health department records over the past 6 years revealed a higher incidence of meningococcal meningitis in blacks due to serogroups Y and W-135 which paralleled the difference in the estimated prevalence of C6 deficiency. Among black residents of the county (n = 235 598) there were 15 cases of meningitis due to these two serogroups, compared with two cases in the white population (n = 422 604) (P = 0.002). We conclude that C6 deficiency is more common among blacks than whites in the south-eastern United States, with a frequency approaching 1 in 1600 black individuals.
Keywords: complement, meningitis, immunodeficiency, genetic mutations
INTRODUCTION
The sixth component of complement, C6, is a 913 amino acid single polypeptide chain serum glycoprotein [1], encoded by a single-copy gene on chromosome 5p13 in proximity to the gene for C7 [2]. C6 is a modular protein and shares structural similarity with the other terminal complement components, C7, C8, and C9, all of which participate in the assembly of the membrane attack complex (MAC) [3]. Formation of the MAC is triggered by cleavage of C5 by a C5 convertase, which is followed by sequential interaction of C5b with C6, C7, C8, and C9. Assembly of the MAC on a cellular membrane results in its gradual insertion into the lipid bilayer and the formation of a transmembrane channel, which can kill susceptible cells or transmit activation or apoptotic signals [4,5].
Genetic deficiencies of all terminal complement components have been described and all have been noted to predispose to neisserial infections, most commonly meningococcal meningitis [6,7]. The susceptibility is attributable to the major role played by complement-mediated killing in host defence against Neisseriae [8] The lifetime risk of neisserial infections in individuals with terminal complement component deficiencies may be as high as 64% [6], although significant variation is noted among different geographical areas and/or ethnic groups [9–11]. For unknown reasons, most studies have shown that strains of Neisseria meningitidis of low pathogenicity, such as serogroups Y and W-135, are over-represented among such patients [6,12,13]. However, other studies failed to demonstrate similar differences in the distribution of serogroups, serotypes, or subtypes among N. meningitidis isolates from complement-deficient and complement-sufficient patients [14,15].
C6 deficiency (C6D), like most other complement protein deficiencies, is inherited as an autosomal recessive trait [15,16]. We have described three different molecular defects leading to C6D, all single nucleotide deletions leading to termination codons (Fig. 1). A 291delC mutation in exon 2 was found in a Japanese individual, while a 1195delC mutation in exon 7 and a 1936delG mutation in exon 12 were found in African-American individuals with C6D [17,18]. Hobart et al. [19] reported a new mutation, 879delG, which is the common defect causing C6D in the Cape Coloured people of South Africa. In preliminary unpublished studies leading up to this report, we identified a new C6 mutation causing C6D: 878delA (described in detail below). This was found in equal frequencies among blacks and whites. Thus, with the exception of 878delA, molecular defects leading to C6D appear to segregate with different racial groups.
Fig. 1.
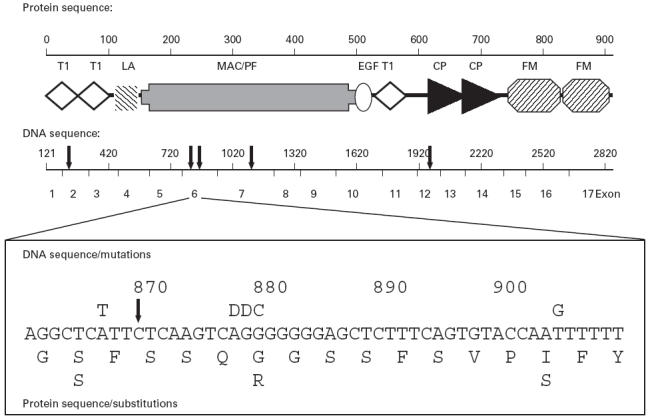
Structure of the human C6 gene and protein showing location of selected mutations. Protein module designation: T1, thrombospondin type 1; LA, LDL receptor class A; MAC/PF, membrane attack complex (MAC)/perforin; EG, epidermal growth factor; CP, complement control protein; FM, Factor I-membrane attack proteins C6/C7. Solid arrows above the DNA sequence indicate deletions described by us previously and in this study and by others (in order from left to right): C291delC [17], 878delA (novel deletion reported here), 879delG [19], 1195delC [18], and 1936delG [17]. The inset shows point mutations and deletions in a region of exon 6. Base substitutions are indicated above the DNA sequence; the corresponding amino acid substitutions are indicated below the peptide sequence. D, Nucleotide deletion.
Population data on the prevalence of C6D are only available for Japan, where it was found to be 2.7 per 100 000 [20]. No data are available for the USA, but studies have indicated a much higher prevalence of the deficiency among individuals of African-American descent than among other ethnic groups [6,21,22]. These observations and our results showing that two of the mutations leading to C6D were found exclusively in African-American kindreds prompted the present study. The results of screening random hospital populations of black and white subjects confirm that 1195delC and 1936delG are limited to African-Americans. More importantly, the data also indicate that C6D is a relatively common genetic disorder among the African-American population of the South-eastern region of the USA.
SUBJECTS AND METHODS
Study subjects
In a protocol approved by the Institutional Review Board, we analysed genomic DNA from 400 individuals (200 black and 200 white) for three molecular defects known to lead to C6D. DNA was isolated from blood samples, which were obtained from the clinical haematology laboratory of the University of Alabama Hospital and were to be otherwise discarded. Blood samples were randomly obtained by screening medical record numbers to prevent duplication and race to obtain equal numbers of samples from black and white patients. Although by design all individuals admitted with N. meningitidis infection were to be excluded from the study, no persons with this diagnosis were found during the acquisition of samples.
Polymerase chain reaction/single-strand conformation polymorphism analysis and nucleotide sequencing
Genomic DNA was isolated from small blood samples (0.5–1 ml) as described [23]. The DNA was extracted twice with phenol/chloroform, dissolved in 100 μl TE buffer (10 mm Tris pH 7.8, 0.01 mm EDTA), and stored at −20°C. Polymerase chain reaction/single-strand conformation polymorphism (PCR/SSCP) was used to screen for three single-base deletions known to cause C6D: the previously unpublished mutations 878delA, 1195delC, and 1936delG, located in exons 6, 7, and 12, respectively. Three primer pairs A, B, and C (pair A: 5′-GTACAAACTGCAGAAGA TGACCT-3′ and 5′-CTTTTTGTGAGAGGCTTGAATGG-3′; pair B: 5′-TGCTAGGTACTTCAACCTTTC-3′ and 5′-GTATTGCAT GCTATCATACCTG-3′; pair C: 5′-GTATGCAATGTGTACACA TGT-3′ and 5′-ACCAAAAGTGAGGTTTAGGAT-3′) were used to amplify exons 6, 7 and 12 of the C6 gene, respectively. The PCR products were labelled with α-32P-dCTP (Amersham, Arlington Heights, IL) as described previously [17]. For SSCP analysis, labelled PCR products were subjected to electrophoresis at room temperature in 6% non-denatured acrylamide gels (Fig. 2). PCR products of exon 7, which were longer than 300 bp, were restricted with Bgl II before SSCP analysis. The single-stranded DNA fragments were visualized by autoradiography. Amplified exons producing aberrant electrophoretic bands were subjected to nucleotide sequencing. Sequencing of PCR products was carried out by using an ABI PRISM 377 DNA Sequencer and the ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction Kit (Perkin Elmer, Foster City, CA), according to the manufacturer's instructions.
Fig. 2.
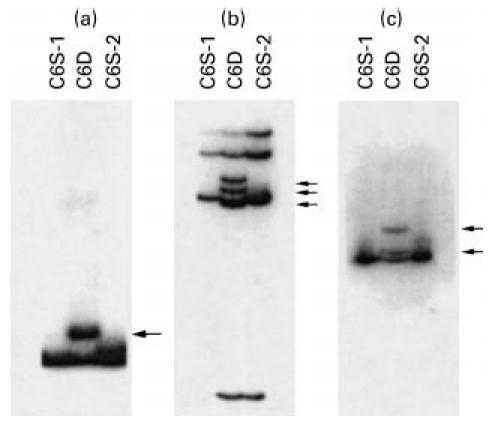
Polymerase chain reaction (PCR)/single strand conformation polymorphism (SSCP) analysis of exon 6 (a), exon 7 (b), and exon 12 (c) of the C6 gene. C6D, Genomic samples from C6-deficient individuals heterozygous for each mutation in the corresponding exon. C6S-1 and C6S-2, samples from unrelated C6-sufficient control subjects. Arrows indicate bands unique to the C6D individuals.
PCR/restriction fragment length polymorphism analysis
Three new point mutations, A867T, G880C, and T905G, were identified in exon 6 during the course of our study. Of these, the G880C mutation creates a novel NspB II restriction site, while the T905G mutation eliminates a Tsp509 I site. The distribution of these two mutations among the study subjects was analysed by PCR/restriction fragment length polymorphism (RFLP). Exon 6 was amplified by PCR, using primer pair A and the 201-bp PCR product was digested with NspB II or Tsp509 I according to the manufacturer's instructions. In the presence of the G880C mutation, the 201-bp PCR product gives rise to two NspB II fragments of 104 and 96 bp, which appear as a single band in 2% agarose gels. In the presence of the T905G mutation, instead of three Tsp509 I fragments of 119, 42 and 40 bp, only two fragments of 159 and 42 bp are generated. It should be noted that the 42-bp and 40-bp fragments are not detected in 2% agarose gels. The A867T mutation was confirmed by nucleotide sequencing the PCR-amplified exon 6 following detection by PCR/SSCP.
Statistical analysis
Data on the prevalence of the three single-base deletional mutations and three novel point mutations of the C6 gene in the two study groups, as well as the incidence of meningitis in Jefferson County among black and white residents, were analysed using Fisher's exact test to determine the probability that the observed differences could have arisen by chance alone.
RESULTS
Identification of a new single-base deletional mutation leading to C6D
In previous investigations of unrelated patients with C6D, the two previously known molecular defects, 1195delC (exon 7), and 1936delG (exon 12), were found exclusively in individuals of African descent. In the course of clinical studies prior to the initiation of the current study protocol, we identified a novel molecular defect in exon 6, consisting of a single-base deletion, 878delA (Fig. 1). The deletion would lead to a shift in the reading frame, generating a novel 45 amino acid sequence, followed by a termination codon in exon 7. The defect was present in two African-American C6D individuals, one homozygous for 878delA and the other a compound heterozygote for 878delA and 1936delG. Neither patient had measurable amounts of C6 protein in serum by radial immunodiffusion, and neither had detectable total (CH50) or C6 haemolytic activity in serum. Both had experienced severe infections with N. meningitidis, one with two episodes of meningococcal meningitis and the other with septic arthritis. Initial analysis of genomic DNA of the two study groups included these three loss-of-function mutations of the C6 gene.
C6D is significantly more common among blacks than whites
Using PCR/SSCP for screening and nucleotide sequencing for confirmation, we found a highly significant difference between the prevalence of two of the three molecular defects (1195delC and 1936delG) among black subjects than among whites (Fig. 2) (Table 1). In fact, among the 200 white individuals tested, we did not find a single carrier of either of these two mutations. In contrast, eight of the 200 black individuals tested were found to be heterozygous for one of the two mutations. This figure projects to a calculated prevalence of C6D (individuals homozygous for one or compound heterozygous for two defects) of 1 in 2500 among the black population of this region of the USA. On the other hand, the newly identified mutation leading to C6D (878delA) was found to be evenly distributed among the two groups. Thus, the prevalence of C6D deficiency due to all three defects can be estimated to be 1 in 1600 blacks compared with approximately 1 in 40 000 whites of this region of the USA.
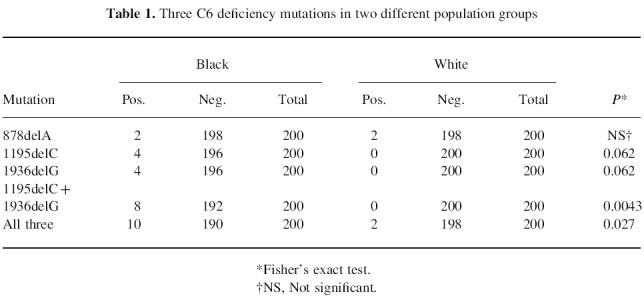
Table 1.
Three C6 deficiency mutations in two different population groups
Prevalence of three newly identified point mutations in exon 6 of the C6 gene
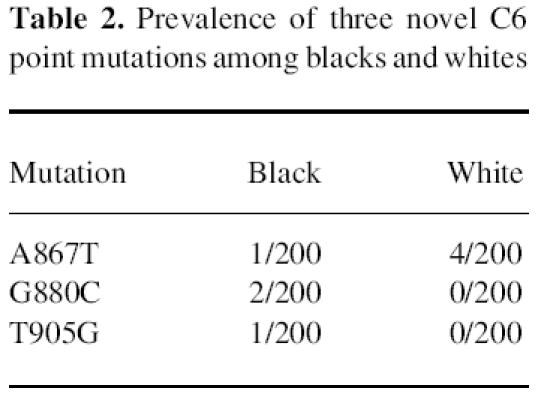
During the course of the study, we identified three new single-base substitutions, all within exon 6, of the C6 gene. These included: (i) an adenine to thymine transversion at position 867, which is silent; (ii) a guanine to cytosine transversion at position 880 which results in replacement of Gly254 by Arg; and (iii) a thymine to guanine transversion at position 905 which results in replacement of IIe262 with Ser. The effects of the latter two point mutations on C6 synthesis, secretion, and function are unknown at present. They both appear to have significant potential to alter C6 serum concentration and/or function, but so far neither has been found in individuals with C6D. The prevalence of these two mutations among the two study groups was analysed by RFLP analysis of the PCR-amplified exon 6 (Fig. 3). The A867T mutation did not create or eliminate a restriction site. For this reason its prevalence in the study groups was determined by nucleotide sequencing of the PCR-amplified exon 6. As shown in Table 2, the prevalence of each of the three novel exon 6 point mutations seems different among blacks and whites, but perhaps because of the small sample size these differences do not reach statistical significance.
Fig. 3.
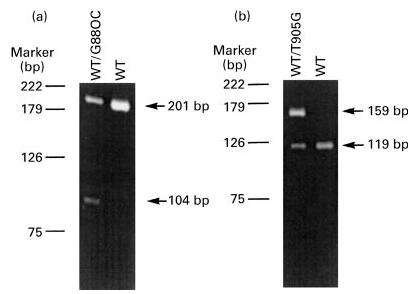
Two exon 6 point mutations (G880C and T905G) detected by polymerase chain reaction (PCR)/restriction fragment length polymorphism (RFLP). (a) A 201-bp DNA fragment was amplified from exon 6 of the C6 gene from two individuals. One of them (WT/G880C) is a heterozygote for the G880C mutation, which creates a restriction site for NspB II resulting in 104-bp and 96-bp fragments that are not resolved in the 2% agarose gel. (b) A 201-bp DNA fragment was amplified from exon 6 of the C6 gene from two subjects. One of them (WT/T905G) is a heterozygote for the T905G mutation, which eliminates a Tsp509 I site giving rise to a 159-bp fragment instead of 119- and 40-bp fragments. The 40- and 42-bp fragments are not detected in the 2% agarose gel.
Table 2.
Prevalence of three novel C6 point mutations among blacks and whites
Increased incidence of meningitis due to unusual serogroups of N. meningitidis in blacks
Individuals with deficiencies of terminal complement components are more susceptible to infection with N. meningitidis. In addition, it has been reported that, at least among certain ethnic groups, meningitis is frequently caused by rare, less pathogenic serogroups such as Y and W-135 [6,12,13]. For the past 6 years, the Jefferson County, Alabama Health Department has collected data on meningococcal meningitis, which include serogroup information. Reporting frequency of this severe disease by area hospitals is virtually 100%. A review of the serogroup information over this 6-year period revealed a strikingly higher incidence of meningococcal meningitis in blacks. This was due mainly to a higher rate of infections with serogroups Y and W-135. There were 29 cases of meningococcal meningitis within a black population of 235 598 over the 6-year period (2.05 cases per 100 000 per year) compared with 21 cases in a white population of 422 604 (0.83 cases/100 000 per year) (Fig. 4; P = 0.002). Total cases due to serogroups B and C numbered 9 and 11 in the black and white populations, respectively (0.64 and 0.43 cases/100 000 per year; P > 0.05), while the combined numbers of infections due to serogroups Y and W135 were 15 and 2, respectively (1.06 and 0.08 per 100 000 per year; P = 0.002).
Fig. 4.
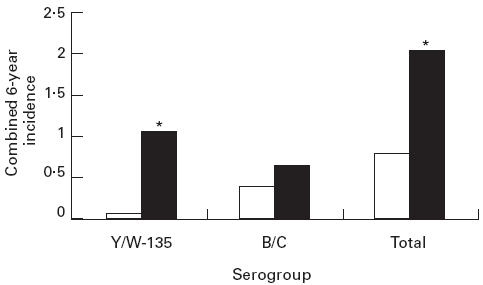
Incidence of meningitis due to different serogroups of Neisseria meningitidis in Jefferson County, AL, over a 6-year period. The data are expressed as cases per 100 000 per year for the cumulative cases over 6 years. *P = 0.002. □, White; ▪, black.
DISCUSSION
The data presented here indicate a surprisingly high prevalence of loss of function molecular defects of the C6 gene among the African-American population of the South-eastern region of the USA. Ten of 200 randomly selected African-Americans had a C6Q0 allele as opposed to two of 200 white subjects. Studies on larger populations are needed to confirm these numbers, but based on the present results, the prevalence of C6D among African-Americans in this region can be calculated to be 0.04%. This is much higher than the 0.0025% prevalence of C6D calculated from our data for the white population of this region and the 0.0027% prevalence of C6D found previously for healthy Japanese blood donors [20]. Therefore, our estimated prevalence of C6D among African-Americans is similar to the most common complement component deficiencies, C9D among Japanese and C2D among Europeans, which are estimated at 0.095% and 0.01%, respectively [24,25]. The epidemiological data on meningococcal meningitis in this County (Fig. 4) are consistent with the estimated higher prevalence of C6D among the African-American population. Over the 6-year period 1992–1997, for which serogroup data were available, the overall infection rate for blacks was 2.5 times higher than for whites and the difference was mainly due to a 13-fold higher incidence of meningococcal meningitis caused by Y and W135 serogroups in blacks than whites. Clearly some of the patients experiencing infection with serogroups Y and W135 may be deficient in another terminal complement component (C7, C8, or C9), as a high incidence of infections due to these rare serogroups has been noted in all of these deficiencies [6,12,13]. However, a higher incidence of terminal component deficiency among African-Americans has been reported mainly for C6 deficiency [6]. We also considered the possibility that the observed difference in infection rates between blacks and whites could be due to socioeconomic factors. However, if that were the case one would have expected that infection rates with the more virulent serogroups of N. meningitidis, B and C, would have been equally high or even higher among blacks.
In their comprehensive review of complement component deficiencies, Ross & Densen [6] list 24 independent kindred comprising C6D individuals. The ethnic origin of 17 of these families, which were derived from different parts of the USA, was known and of these 11 were of African-American descent. Thus, it seems unlikely that the observed high rate of C6D is confined to the African-American population of the South-eastern USA. Four of the 10 African-American C6D heterozygotes of the present study had the 1195delC and another four the 1936delG mutation, as opposed to none of the two white heterozygotes. The 1195delC mutation was initially described in an African-American family from this area [18]. We subsequently detected the same mutation in three additional African-American kindreds from different parts of the USA, including the North-east and the Midwest (unpublished). The same mutation was also found in two unrelated black South Africans, a British subject of African Caribbean descent, and two Cape Coloured families [19]. Cape Coloured people are of mixed descent and their gene pool is considered to have been derived equally from Caucasoid, South-East Asian, and South African (Bushman, Hottentot, and black African) populations. The 1936delG defect was also originally described in an African-American family from this region [17]. It was subsequently found in four additional unrelated C6D African-American families from this and other regions of the USA (unpublished). This mutation was also detected in one of the Cape Coloured families studied by Hobart et al., as well as in the individual of African Caribbean descent they studied [19]. Therefore, it appears likely that these two loss of function mutations of the C6 gene originated in West Africa among populations that were subsequently moved to other parts of the world through the slave trade. The high prevalence observed in this study and the wide distribution of these two defects suggest their high prevalence in the original African population. Both of these mutations are single-base deletions, which represent less than 10% of human mutations and are more likely to lead to loss of function than point mutations, which are much more common events [26].
In contrast to 1195delC and 1936delG, the third mutation included in our screening, 878delA, was found equally distributed among blacks and whites. This is a new loss of function molecular defect, and was included in the study because it was originally discovered in two unrelated C6D African-American individuals. It is of interest that A878 immediately precedes a string of seven Gs, one of which is deleted in the mutation reported by Hobart et al. to predominate among C6D Cape Coloured people [19]. By convention that deletion was assigned to the first G, at position 879 (Fig. 1). The 879delG mutation was also found in two Dutch families, but for several reasons the authors favoured the view that the two mutations probably arose independently. In addition to these deletional mutations in exon 6, we detected three point mutations, A867T, G880C, and T905G. Two of those result in amino acid substitutions and could lead to loss of C6 function. The three point mutations are within 20 nucleotides upstream or downstream of the string of seven Gs. The presence of five or perhaps six mutations within 40 nucleotides of an exon is unusual and suggests a strong mutagenic mechanism, possibly related to misalignment during replication caused by the seven tandemly repeated Gs.
The high prevalence of loss of function mutations among African-Americans and presumably other Africans as well raises the question of a selective advantage associated with C6D. This possibility was also entertained by other investigators [2,7] and was supported by the finding of an excess of homozygous deficient siblings over the expected number in families of C6D probands [7]. Given the susceptibility to meningococcal infections of homozygous C6D individuals, arguments for a selective advantage appear to be counterintuitive. The 5–10 000-fold higher frequency of meningitis in C6D subjects is however counterbalanced to some extent by a milder clinical picture and a 6.5-fold lower mortality rate compared with C6-sufficient individuals [6]. These clinical findings are partly explained by studies demonstrating the release of endotoxin from Gram-negative bacteria by the insertion of the MAC into the bacterial membrane [27,28]. Inability to form the MAC in C6D will result in lower levels of endotoxin and a more favourable clinical course [29]. It has been argued that this beneficial effect provides the selective advantage, as it should be expected to occur not only in meningitis but also in other Gram-negative infections to which complement-deficient individuals have no increased susceptibility [7].
Acknowledgments
The authors express their gratitude to Dr Richard Whitley for referral of a patient and to John Bennett for technical assistance. This work has been supported in part by AR44505 (J.E.V.) and a Merit Review Award from the Department of Veteran's Affairs, and by Public Health Service grant R01-AI20880 from the National Institute of Allergy and Infectious Disease (P.D.).
REFERENCES
- 1.DiScipio RG, Hugli TE. The molecular architecture of human complement component C6. J Biol Chem. 1989;264:16197–206. [PubMed] [Google Scholar]
- 2.Hobart MJ, Fernie BA, DiScipio RG, Lachmann PJ. A physical map of the C6 and C7 complement component gene region on chromosome 5p13. Hum Mol Genet. 1993;2:1035–6. doi: 10.1093/hmg/2.7.1035. [DOI] [PubMed] [Google Scholar]
- 3.Plumb ME, Sodetz JM. Proteins of the membrane attack complex. In: Volanakis JE, Frank MM, editors. The human complement system in health and disease. New York: Marcel Dekker, Inc.; 1998. pp. 49–82. [Google Scholar]
- 4.Mueller-Eberhard HJ. The membrane attack complex of complement. Annu Rev Immunol. 1986;4:503–28. doi: 10.1146/annurev.iy.04.040186.002443. [DOI] [PubMed] [Google Scholar]
- 5.Niculescu F, Rus H, van Biesen T, Shin ML. Activation of Ras and mitogen-activated protein kinase pathway by terminal complement complexes is G protein dependent. J Immunol. 1997;158:4405–12. [PubMed] [Google Scholar]
- 6.Ross SC, Densen P. Complement deficiency states and infection: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine. 1984;63:243–73. [PubMed] [Google Scholar]
- 7.Orren A, Potter PC, Cooper RC, Du Toit E. Deficiency of the sixth component of complement and susceptibility to Neisseria meningitidis infections: studies in 10 families and five isolated cases. Immunology. 1987;62:249–53. [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholson A, Lepow IH. Host defense against Neisseria meningitidis requires a complement-dependent bactericidal activity. Science. 1979;205:298–9. doi: 10.1126/science.451601. [DOI] [PubMed] [Google Scholar]
- 9.Platonov AE, Beloborodov VB, Vershinina IV. Meningococcal disease in patients with late complement component deficiency: studies in the U.S.S.R. Medicine. 1993;72:374–445. doi: 10.1097/00005792-199311000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen HE, Koch C, Magnussen P, Lind I. Complement deficiencies in selected groups of patients with meningococcal disease. Scand J Infect Dis. 1989;21:389–96. doi: 10.3109/00365548909167442. [DOI] [PubMed] [Google Scholar]
- 11.Moller Rasmussen J, Brandslund I, Teisner B, et al. Screening for complement deficiencies in unselected patients with meningitis. Clin Exp Immunol. 1987;68:437–45. [PMC free article] [PubMed] [Google Scholar]
- 12.Fijen CAP, Kuijper EJ, Hannema AJ, Sjoholm AG, van Putten JPM. Complement deficiencies in patients over ten years old with meningococcal disease due to uncommon serogroups. Lancet. 1989;2:585–8. doi: 10.1016/s0140-6736(89)90712-5. [DOI] [PubMed] [Google Scholar]
- 13.Mayatepek E, Grauer M, Hansh GM, Sonntag HG. Deafness, complement deficiencies and immunoglobulin status in patients with meningococcal diseases due to uncommon serogroups. Pediatr Infect Dis J. 1993;12:808–11. doi: 10.1097/00006454-199310000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Orren A, Caugant DA, Fijen CAP, Dankert J, van Schalkwyk EJ, Poolman JT, Coetzee GJ. Characterization of strains of Neisseria meningitidis recovered from complement-sufficient and complement-deficient patients in the Western Cape Province, South Africa. J Clin Microbiol. 1994;32:2185–91. doi: 10.1128/jcm.32.9.2185-2191.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wurzner R, Witzel-Schlomp K, Tokunaga K, Fernie BA, Hobart MJ, Orren A. Reference typing report for complement components C6, C7 and C9 including mutations leading to deficiencies. Exp Clin Immunogenet. 1998;15:268–85. doi: 10.1159/000019082. [DOI] [PubMed] [Google Scholar]
- 16.Colten HR, Rosen FS. Complement deficiencies. Annu Rev Immunol. 1992;10:809–34. doi: 10.1146/annurev.iy.10.040192.004113. [DOI] [PubMed] [Google Scholar]
- 17.Nishizaka H, Horiuchi T, Zhu Z-B, et al. Molecular bases for inherited human complement component C6 deficiency in two unrelated individuals. J Immunol. 1996;156:2309–15. [PubMed] [Google Scholar]
- 18.Zhu Z-B, Totemchokchyakarn K, Atkinson TP, Volanakis JE. Molecular defects leading to human complement component C6 deficiency in an African-American family. Clin Exp Immunol. 1998;111:91–6. doi: 10.1046/j.1365-2249.1998.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hobart MJ, Fernie BA, Fijen KAPM, Orren A. The molecular bases of C6 deficiency in the Western Cape, South Africa. Hum Genet. 1998;103:506–12. doi: 10.1007/s004390050858. [DOI] [PubMed] [Google Scholar]
- 20.Inai S, Akagaki Y, Moriyama T, Fukumori Y, Yoshimura K, Ohnoki S, Yamaguchi H. Inherited deficiencies of late-acting complement components other than C9 found among healthy blood donors. Int Arch Allergy Appl Immunol. 1989;90:274–9. doi: 10.1159/000235037. [DOI] [PubMed] [Google Scholar]
- 21.Ellison RT, III, Kohler PF, Curd JG, Judson FN, Reller LB. Prevalence of congenital or acquired complement deficiency in patients with sporadic meningococcal disease. N Engl J Med. 1983;308:913–6. doi: 10.1056/NEJM198304213081601. [DOI] [PubMed] [Google Scholar]
- 22.Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. 1991;4:359–95. doi: 10.1128/cmr.4.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee TH, Donegan E, Slichter S, Busch MP. Transient increase in circulating donor leukocytes after allogeneic transfusions in immunocompetent recipients compatible with donor cell proliferation. Blood. 1995;85:1207–14. [PubMed] [Google Scholar]
- 24.Rynes RI, Britten AFH, Pickering RJ. Deficiency of the second complement component: association with the HLA haplotype A10, B18 in a normal population. Ann Rheum Dis. 1982;41:93. doi: 10.1136/ard.41.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukumori Y, Yoshimura K, Ohnoki S, Yamaguchi H, Akagaki Y, Inai S. A high incidence of C9 deficiency among healthy blood donors in Osaka. Japan Int Immunol. 1989;1:85–9. doi: 10.1093/intimm/1.1.85. [DOI] [PubMed] [Google Scholar]
- 26.Cooper DN, Krawczak M, Antonarakis SE. The nature and mechanisms of human gene mutation. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 7. New York: McGraw-Hill, Inc.; 1995. pp. 259–91. [Google Scholar]
- 27.Brandtzaeg P, Mollnes TE, Kierulf P. Complement activities and endotoxin levels in systemic meningococcal disease. J Infect Dis. 1989;160:58–65. doi: 10.1093/infdis/160.1.58. [DOI] [PubMed] [Google Scholar]
- 28.Lehner PJ, Davies KA, Walport MJ, Cope AP, Wuerzner R, Orren A, Morgan BP, Cohen J. Meningococcal septicaemia in a C6 deficient patient and effects of plasma transfusion on lipopolysaccharide release. Lancet. 1992;2:1379–81. doi: 10.1016/0140-6736(92)92561-s. [DOI] [PubMed] [Google Scholar]
- 29.Lachmann PJ. Complement—friend or foe? Br J Rheumatol. 1987;26:409–15. doi: 10.1093/rheumatology/26.6.409. [DOI] [PubMed] [Google Scholar]