

Abstract
Crohn's disease (CD) is a multifactorial disease with genetic heterogeneity. TNF-α plays a key role in the development of the mucosal lesions. The aim of our work was to study a single base pair polymorphism located in the promoter region of TNF gene, in a large population of CD patients with well defined phenotypes. One hundred and ninety-three patients with CD and 98 ethnically matched controls were studied. The −308 single base pair polymorphism of TNF gene was studied using an allele-specific polymerase chain reaction. Genotype and allelic frequencies were compared between patients and controls and between subgroups of patients defined by sex, age at diagnosis, familial history, location of disease, type of disease, extra-intestinal manifestations, and response to steroid treatment. In 29 patients a measure of TNF-α production by colonic biopsies was performed. The frequency of the allele TNF2 as well as the proportion of carriers of the allele TNF2 were slightly but not significantly lower in CD than in controls (11.9% versus 14.8% and 21.5% versus 27.6%, respectively). A more prominent difference in frequencies of allele TNF2 and in proportions of TNF2 carriers was found when comparing subgroups of patients. The frequency of allele TNF2 was significantly higher in steroid-dependent than in non-steroid-dependent disease (28.1% versus 10.3%; Δ = 17.8%, 95% confidence interval (CI) = 6.3–29.5%, P = 0.0027) and tended to be higher in colonic than in small bowel disease and in fistulizing than in stricturing disease. Furthermore, TNF2 carriers tended to be more frequent in patients with steroid-dependent than non-steroid-dependent disease (43.8% versus 19.3%; Δ = 24.5%, 95% CI = 3.6–45.4%, P = 0.022), in patients with fistulizing than stricturing disease (26.5% versus 9.6%; Δ = 16.9%, 95% CI = 1.1–32.6%, P = 0.036), and in patients with colonic than small bowel disease (26.5% versus 11.1%; Δ = 15.4%, 95% CI = −0.8–31.6%, P = 0.063). Finally, patients carrying at least one copy of allele 2 were found to produce slightly more TNF-α at the colonic level. The −308 TNF gene polymorphism may have a slight influence on the behaviour of CD. The carriage of allele 2 may favour steroid-dependent disease and to a lesser extent fistulizing and colonic disease, possibly secondary to a more intense TNF-α-driven inflammatory reaction at the mucosal level.
Keywords: Crohn's disease, genetics, tumour necrosis factor-alpha
INTRODUCTION
In Crohn's disease (CD), a genetic predisposition was first suggested by epidemiological studies [1]. The most convincing data come from twin studies, showing a higher concordance rate for the disease among monozygotic than dizygotic twins [2]. More recently, wide genome screenings have confirmed a genetic predisposition and have identified regions significantly associated with the transmission of the disease on chromosomes 16 and 12 [3,4].
Clinical and epidemiological data suggest that CD is a heterogeneous entity and familial studies have shown a relative conservation of the disease phenotype among affected members in the same family [5,6]. The genetic background may therefore not only explain the predisposition to the disease but also influence its phenotype. A few preliminary studies have shown an association between a genetic polymorphism and the phenotype of CD [7–9]. The genes potentially influencing the phenotype of the disease may be different from the ones responsible for the predisposition to the disease and may be missed by the wide genome screenings. The TNF gene located on chromosome 6 in the HLA region is a good candidate gene to look for genotype–phenotype relationships. Indeed, the production of TNF-α is increased in the mucosa of patients with CD [10,11] and anti-TNF therapies have shown dramatic effects on the activity of the disease [12,13]. Several polymorphisms have been described in the TNF gene and some of them have already been studied in CD [14–18]. However, these studies looked for genes involved in the overall predisposition to the disease and did not study carefully the genotype–phenotype correlations. The single base pair (SBP) polymorphism at position −308 in the promoter of TNF-α gene has been shown to have a functional significance on TNF-α production [19–21].
The aim of our work was to study the TNF gene −308 SBP polymorphism in various subgroups of CD patients defined by classical phenotypes. We also looked at the production of TNF-α by the colonic mucosa of CD patients according to their genotypes.
SUBJECTS AND METHODS
Subjects
Ethical approval for this study was given by the Liège Ethics Committee in March 1995. Clinical data were obtained by review of case records for all patients. Patients were selected at random from adult out-patients attending the gastroenterology clinics at the University Hospital of Liège, at the St Joseph Hospital, Liège, and at University Hospital of Leuven. When a patient had a family history of CD, no other affected members of the family were recruited for the present study. The diagnosis of CD was made on the basis of clinical, radiological and histological data, according to standard criteria. Disease phenotypes were defined by sex, age at diagnosis, familial disease, disease location, type of disease, extra-intestinal manifestations, response to steroid. Patients were categorized in seven subgroups, according to the age at diagnosis (0–20, 21–30, 31–40, 41–50, 51–60, 61–70 and 71–80 years). Disease extent was defined on the basis of a combination of clinical, radiological, histological, and endoscopic evidence. The type of disease was defined as predominantly stricturing, fistulizing or inflammatory. Steroid dependency was defined by either two successive relapses during steroid tapering or two successive relapses occurring early (within 2 months) after steroid discontinuation.
CD patients
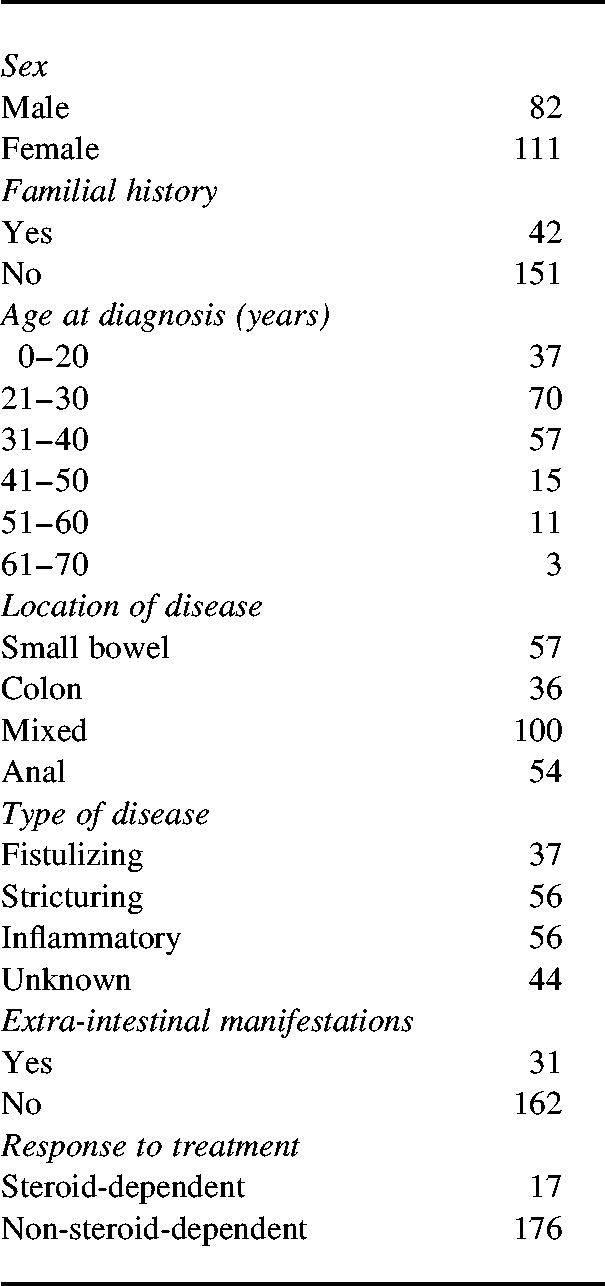
One hundred and ninety-three unrelated patients with definite CD were studied. Characteristics of the patients are shown in Table 1.
Table 1.
Characteristics of patients
Controls
Ninety-eight unrelated European Caucasoid individuals served as controls. These control subjects were either hospital workers or prospective blood donors. None had a personal or familial history of inflammatory bowel disease (IBD).
DNA extraction
Genomic DNA was extracted from 10 ml venous blood using a modified salting out technique [22] and resuspended in sterile distilled water at a final concentration of 0.1–1.0 μg/μl, before use.
TNF −308 SBP substitution
The substitution was studied by a polymerase chain reaction (PCR) involving primers specific for each allele of the G to A polymorphism at residue −308, as previously described [16]. In 177 patients, samples were successfully genotyped; in 16 samples, amplification was unsuccessful (either because of inadequate DNA, or non-specific amplification).
After amplification, DNA samples were electrophoresed in 1% agarose gels containing ethidium bromide, and visualized under ultraviolet light.
Culture of colonic biopsies
Colonic biopsy specimens were obtained from macroscopically and microscopically uninflamed (n = 24) or inflamed (n = 20) areas during colonoscopy in 29 patients with IBD. All patients required colonoscopy for medical reasons. For each patient a set of five biopsies in inflamed and/or uninflamed areas was taken. Two were fixed in formalin for histological assessment, three were placed at 4°C in a medium consisting of Ca2+- and Mg2+-free (CMF) Hanks' solution (Gibco, Life Technologies, Paisley, UK), supplemented by 100 U/ml penicillin and 100 μg/ml streptomycin.
Tissue culture
After collection, biopsy specimens were transferred to the laboratory and cultured as previously described [23]. After 18 h culture at 37°C in a humidified 95% air/5% CO2 atmosphere, medium was removed, centrifuged at 1500 g for 5 min and stored at −80°C until required for cytokine assays. Structural integrity was assessed by standard histology and by measurement of lactate dehydrogenase release according to Wardle et al. [24]. After 18 h culture, no histological changes were noted compared with precultured tissue. Furthermore, the release of lactate dehydrogenase in cultured tissues was significantly lower than in uncultured tissues (data not shown).
Immunoassays for cytokines
TNF-α and IL-6 production was measured using specific immunoassays (EASIA; Biosources Europe, Fleurus, Belgium). Immunoassays were performed according to the manufacturer's instructions. The detection limit was 3 pg/ml for TNF-α and 2 pg/ml for IL-6. TNF-α production was expressed by its concentration per ml of culture medium and per mg of tissue in culture.
The production of TNF-α at the mucosal level may be influenced by the degree of inflammation. This may interfere with a possible genetic influence. We thus expressed TNF production by the ratio between TNF-α concentration in the supernatants of colonic biopsies and histological score of inflammation. The overall inflammation score was adapted from Riley et al. [25], as previously described [23]. According to Reimund et al. histological inflammation correlates well with IL-6 production by tissue culture [23]. So, to take into account more accurately the variations in the degree of inflammation in the assessment of individual colonic TNF-α production, we also expressed TNF-α production as a ratio between TNF-α and IL-6 concentrations (units of TNF-α production) in the supernatants of biopsy cultures. The final result for each patient corresponds to the mean of three individual biopsy cultures in inflamed and/or uninflamed regions.
Statistical analysis
Groups and subgroups of patients were compared on the basis of genotype and allelic frequencies. Differences in genotype proportions and allelic frequencies were assessed by Student's t-test and the results are given as a difference in proportion of genotypes or allelic frequencies (Δ) with a 95% confidence interval (CI) and a P value. For subgroup comparisons, as multiple tests were performed, a more conservative level of significance for P value was taken (P = 0.005).
The results of TNF-α production by colonic biopsies are expressed as median and interquartile ranges. The comparison between groups according to the −308 TNF genotype was made using Mann–Whitney U-test. The level of significance was taken as P < 0.05.
RESULTS
TNF polymorphism
Thirty-eight (21.5%) patients and 27 (27.6%) healthy subjects were carriers of at least one copy of allele 2 (Δ = 6.1%; 95% CI = −4.4–16.6%, P = 0.26).Those include two healthy subjects (2%) and four patients with CD (2.3%) who were homozygous for allele 2. The frequencies of the allele TNF2 were 11.9% in CD and 14.8% in controls (Δ = 2.9%; 95% CI = −2.9–8.7%, P = 0.32).
On subgroup analysis, the only significant difference was when comparing allelic frequencies between steroid-dependent and non-steroid-dependent patients (Δ = 17.8%; 95% CI = 6.3–29.5%, P = 0.0027). The proportion of TNF2 carriers was slightly higher, but without reaching the conservative P value of 0.005, in steroid-dependent than in non-steroid-dependent disease (Δ = 24.6%; 95% CI = 3.6–45.4%, P = 0.022), in fistulizing than stricturing disease (Δ = 16.9%; 95% CI = 1.1–32.6%, P = 0.036) and in colonic than small bowel disease (Δ = 15.4%; 95% CI = −0.8–31.6%, P = 0.063).
Allelic frequencies as well as frequencies of TNF1 homozygotes and TNF2 carriers in these subgroups of patients are shown in Table 2.
Table 2.
TNF gene polymorphism in various phenotypes of Crohn's disease (CD): allelic frequencies (%) and proportions of genotypes (%)

Genotypes and allelic frequencies were similar in subgroups of patients defined by sex, age at diagnosis, extra-intestinal manifestations or familial history.
TNF-α production depending on the −308 TNF genotype
Among the 29 patients who had a measure of TNF-α production by colonic biopsies, eight were carriers of at least one copy of allele 2 (including two TNF2 homozygotes). When the TNF-α production was corrected by a histological score of inflammation, there was no significant difference between TNF2 carriers and TNF1 homozygotes, either in inflamed (18.2 pg/ml.mg (9.9–26.4) versus 6.4 pg/ml.mg (3.4–18.3), respectively; P = 0.18) or in uninflamed areas (5.4 pg/ml.mg (4.8–6.0) versus 3.9 pg/ml.mg (1.4–7.0), respectively; P = 0.19). When TNF-α production was corrected by the production of IL-6 by the same biopsy, TNF-α production in inflamed regions tended to be higher among the carriers of allele 2 (n = 8) compared with TNF1 homozygotes (n = 12) (0.07 (0.035–0.095) and 0.032 (0.018–0.060), respectively; P = 0.069) (Fig. 1). In uninflamed regions, there was no significant difference between carriers of allele 2 (n = 5) and TNF1 homozygotes (n = 19) (0.039 (0.037–0.055) and 0.078 (0.031–0.148), respectively).
Fig. 1.
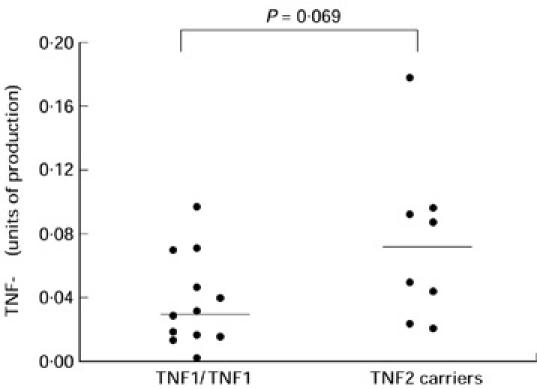
TNF-α production by colonic biopsies taken in inflamed areas, depending on the −308 TNF genotype. Biopsies were cultured for 18 h. TNF-α concentrations were measured on the supernatants by immunoassay. TNF-α production is expressed as a ratio between TNF-α and IL-6 concentrations in the same supernatants to correct for the degree of inflammation (units of TNF-α production). Each point corresponds to the mean of measures performed on three biopsy cultures in the same patient. The production in TNF1 homozygotes (n = 12) and TNF2 carriers (n = 8) was 0.032 (0.018–0.060) and 0.070 (0.035–0.095), respectively (median (interquartile ranges)) (P = 0.069).
DISCUSSION
We have shown differences, some statistically significant, in allelic frequencies as well as in proportions of genotypes for the −308 SBP substitution in the promoter of TNF gene, in various subgroups of CD patients characterized by different clinical phenotypes. Moreover, the carriage of at least one copy of allele 2 was associated with a tendency to produce more TNF-α in the inflamed mucosa at the colonic level.
Previous studies of the polymorphism of TNF gene in CD have produced controversial results. Some studies have suggested significant associations between CD and TNF gene polymorphisms or haplotypes [16,17] that were not confirmed for other populations of patients [14,15,18], suggesting possible differences between various ethnic groups or between various subgroups of CD. In the present study an overall slight decrease of the allele TNF2 was shown in CD. However, this was not statistically significant. The decrease in TNF2 frequency was more prominent in subgroups of patients, including isolated small bowel disease and stricturing disease. This emphasizes that some of the differences reported in the previous studies of TNF gene polymorphisms in CD [14–18] may partly be due to different proportions of CD phenotypes represented.
In contrast, a significant increase of TNF2 allele frequency was found in patients with steroid-dependent CD. Albeit found in a small sample of patients (n = 16) and although it was not associated with a significant increase in the proportion of patients carrying the allele TNF2 (probably due to the small size of the sample), this increase seems worth mentioning. The mechanisms of steroid dependency in CD are unknown. As it has been documented in other situations, it may reflect either a primary phenomenon [26] or a secondary phenomenon linked to the intensity of the inflammation [27]. An increased TNF2 frequency has been described in several infectious diseases associated with high production of TNF-α [28,29]. Moreover, TNF2 allele has been associated with increased TNF-α transcription [19] and also production by isolated peripheral blood mononuclear cells [20] as well as in a whole blood cell culture model [21], although these associations were not always found [30,31]. In the present study we show preliminary data suggesting also an association between the carriage of TNF2 allele and a slightly increased production of TNF-α at the colonic level in inflamed regions. The association between steroid-dependent CD and TNF2 allele may thus be due to an increased capacity for TNF-α production at the colonic level giving rise to more inflammation and possibly to a secondary steroid dependency. Alternatively, the association between steroid-dependent CD and TNF2 allele may reflect an association with another gene polymorphism in linkage disequilibrium with TNF2. Interestingly, an association has also been reported between a TAP transporter gene polymorphism, located close to TNF gene, and the response to steroid treatment in CD [9].
Although not statistically significant, we also found an increased frequency of allele TNF2 as well as an increased carriage rate of TNF2 in patients with fistulizing disease. In contrast, a recent study in a Dutch population [7] showed a negative association between fistulizing CD and the allele HLA-DRB1*03 classically in linkage disequilibrium with TNF2 [16,32]. However, interestingly, in that population there was no decrease in TNF2 frequency, suggesting recombination between DRB1 and TNF-α loci in patients with fistulizing CD. As for steroid-dependent CD, this association with a particular pathological type of CD may reflect a more aggressive inflammatory reaction linked to the magnitude of TNF-α production. Alternatively, as has been suggested in leprosy [33], this association may reflect a particular pathway of the immune response, independently of its intensity.
There was also a slight increase in the frequency of allele TNF2 and in TNF2 carriage rate in colonic disease compared with small bowel disease. If this difference reflects a true association, it may suggest a different implication of TNF-α in lesions of CD in the small bowel or the colon. No data comparing TNF production at the mucosal level in the colon and small intestine are available, but differences in intraluminal material in vivo at these two places could determine different profiles of local immunological reaction.
It is important to emphasize that all these associations between TNF2 allele and either phenotypes of CD or TNF-α production in inflamed mucosa are slight, borderline or even not statistically significant. This suggests that beside TNF gene, other genetic or environmental factors are involved in the determination of these biological or clinical parameters.
In conclusion, although TNF gene polymorphism is probably not critical in overall susceptibility to CD, this study suggests that it may play a role in the behaviour of the disease. The carriage of the allele TNF2 of the −308 SBP polymorphism seems to be associated with steroid-dependent disease and to a lesser extent with fistulizing and colonic disease.
Acknowledgments
E.L. is supported by a grant from the National Fund for Scientific Research, Belgium. The work was supported by a grant from the University Hospital of Liège.
REFERENCES
- 1.Satsangi J, Jewell DP, Rosenberg WMC, Bell JI. Genetics of inflammatory bowel disease. Gut. 1994;35:696–700. doi: 10.1136/gut.35.5.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tysk C, Lindberg E, Järnerot G, Floderus-Myrhed B. Ulcerative colitis and Crohn's disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut. 1988;28:990–6. doi: 10.1136/gut.29.7.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hugot JP, Laurent-Puig P, Gower-Rousseau C, et al. Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature. 1996;379:821–3. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 4.Satsangi J, Parkes M, Louis E, et al. Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nature Genetics. 1996;14:199–202. doi: 10.1038/ng1096-199. [DOI] [PubMed] [Google Scholar]
- 5.Peeters M, Nevens H, Baert F, Hiele M, De Meyer AM, Vlietinck R, Rutgeerts P. Familial aggregation in Crohn's disease: increased aged-adjusted risk and concordance in clinical characteristics. Gastroenterology. 1996;111:597–603. doi: 10.1053/gast.1996.v111.pm8780562. [DOI] [PubMed] [Google Scholar]
- 6.Colombel JF, Grandbastien B, Gower-Rousseau C, et al. Clinical charateristics of Crohn's disease in 72 families. Gastroenterology. 1996;111:604–7. doi: 10.1053/gast.1996.v111.pm8780563. [DOI] [PubMed] [Google Scholar]
- 7.Bouma G, Poen AC, Garcia-Gonzalez MA, Schreuder GMT, Felt-Bersma RJF, Meuwissen SGM, Pena AS. HLA-DRB1*03, but not the TNFA-308 promoter gene polymorphism, confers protection against fistulising Crohn's disease. Immunogenetics. 1998;47:451–5. doi: 10.1007/s002510050382. [DOI] [PubMed] [Google Scholar]
- 8.Plevy SE, Taylor K, De Woody KL, Schaible TF, Shealy D, Targan SR. Tumor necrosis factor (TNF) microsatellite haplotypes and perinuclear anti-neutrophil cytoplasmic antibody (pANCA) identify Crohn's disease (CD) patients with poor clinical response to anti-TNF monoclonal antibody (cA2) Gastroenterology. 1997;112:A1062. [Google Scholar]
- 9.Heresbach D, Alizadeh M, Bretagne JF, et al. TAP gene transporter polymorphism in inflammatory bowel diseases. Scand J Gastroenterol. 1997;32:1022–7. doi: 10.3109/00365529709011219. [DOI] [PubMed] [Google Scholar]
- 10.Dionne S, Hiscott J, D'agata I, Duhaime A, Seidman EG. Quantitative PCR analysis of TNF-α and IL-1β mRNA levels in pediatric IBD mucosal biopsies. Dig Dis Sci. 1997;42:1557–66. doi: 10.1023/a:1018895500721. [DOI] [PubMed] [Google Scholar]
- 11.Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP. Enhanced secretion of tumor necrosis factor-alpha, IL-6 and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1993;94:174–81. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stack WA, Mann SD, Roy AJ, et al. Randomised controlled trial of CDP571 antibody to tumour necrosis factor-α in Crohn's disease. Lancet. 1997;349:521–4. doi: 10.1016/s0140-6736(97)80083-9. [DOI] [PubMed] [Google Scholar]
- 13.Targan SR, Hanauer SB, Van Deventer SJH, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor α for Crohn's disease. N Engl J Med. 1997;337:1029–35. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 14.Mansfield JC, Holden H, Tarlow JK, Di Giovine FS, McDowell TL, Wilson AG, Holdsworth CD, Duff GW. Novel genetic association between ulcerative colitis and the anti-inflammatory cytokine interleukin-1 receptor antagonist. Gastroenterology. 1994;106:637–42. doi: 10.1016/0016-5085(94)90696-3. [DOI] [PubMed] [Google Scholar]
- 15.Bouma G, Xia B, Crusius JBA, Bioque G, Koutroubakis I, Von Blomberg VME, Meuwissen SGM, Pena AS. Distribution of four polymorphisms in the tumor necrosis factor (TNF) genes in patients with inflammatory bowel disease (IBD) Clin Exp Immunol. 1996;103:391–6. doi: 10.1111/j.1365-2249.1996.tb08292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louis E, Satsangi J, Roussomoustakaki M, Parkes M, Fanning G, Welsh K, Jewell DP. Cytokine gene polymorphisms in inflammatory bowel disease. Gut. 1996;39:705–10. doi: 10.1136/gut.39.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plevy SE, Targan SR, Yang H, Fernandez D, Rotter JI, Toyoda H. Tumor necrosis factor microsatellites define a Crohn's disease-associated haplotype on chromosome 6. Gastroenterology. 1996;110:1053–60. doi: 10.1053/gast.1996.v110.pm8612993. [DOI] [PubMed] [Google Scholar]
- 18.Heresbach D, Ababou A, Bourienne A, et al. Tumor necrosis factor gene and microsatellite polymorphism in chronic inflammatory bowel diseases. Gastroenterol Clin Biol. 1997;21:555–61. [PubMed] [Google Scholar]
- 19.Wilson AG, Symons JA, McDowel TL, McDevitt HO, Duff GW. Effect of a polymorphism in the human tumor necrosis factor α promoter on transcriptional activation. Proc Natl Acad Sci USA. 1997;94:3195–9. doi: 10.1073/pnas.94.7.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouma G, Crusius JBA, Oudkerk Pool M, et al. Secretion of tumor necrosis factor α and lymphotoxin α in relation to polymorphisms in the TNF genes and HLA-DR alleles. Relevance for inflammatory bowel disease. Scand J Immunol. 1996;43:456–63. doi: 10.1046/j.1365-3083.1996.d01-65.x. [DOI] [PubMed] [Google Scholar]
- 21.Louis E, Franchimont D, Piron A, et al. Tumor necrosis factor gene polymorphism influences tumor necrosis factor α production in LPS-stimulated whole blood cell culture in healthy humans. Clin Exp Immunol. 1998;113:401–6. doi: 10.1046/j.1365-2249.1998.00662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bunce M, Taylor CJ, Welsh KI. Rapid HLA-DQB typing by eight PCR amplifications with sequence-specific primers (PCR-SSP) Human Immunol. 1993;37:201–6. doi: 10.1016/0198-8859(93)90502-r. [DOI] [PubMed] [Google Scholar]
- 23.Reimund JM, Wittersheim C, Dumont S, Muller CD, Kenney JS, Baumann R, Poindron P, Duclos B. Increased production of tumor necrosis factor-α, interleukin-1β and interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn's disease. Gut. 1996;39:684–9. doi: 10.1136/gut.39.5.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wardle TD, Hall L, Turnberg LA. Use of coculture of colonic mucosal biopsies to investigate the release of eicosanoids by inflamed and uninflamed mucosa from patients with inflammatory bowel disease. Gut. 1992;33:1644–51. doi: 10.1136/gut.33.12.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riley SA, Mani V, Goodman MJ, Dutt S, Herd ME. Microscopic activity in ulcerative colitis: what does it mean ? Gut. 1991;32:174–8. doi: 10.1136/gut.32.2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sher ER, Leung DYM, Surs W, Kam JC, Zieg G, Kamada AK, Harbeck R, Szefler SJ. Steroid resistant asthma: cellular mechanisms contributing to inadequate response to glucocorticoid therapy. J Clin Invest. 1994;93:33–9. doi: 10.1172/JCI116963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van der Burg B, Liden J, Okret S, Delaunay F, Wissink S, Van der Saag PT, Gustafsson JA. Nuclear factor-κB repression in anti-inflammation and immunosuppression by glucocorticoids. Trends Endocrinol Metab. 1997;8:152–7. doi: 10.1016/s1043-2760(97)00006-4. [DOI] [PubMed] [Google Scholar]
- 28.McGuire W, Hill AVS, Allsopp CEM, Greenwood BM, Kiatkowski D. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature. 1994;371:508–11. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 29.Conway DJ, Holand MJ, Bailey RL, Campbell AE, Mahdi OMS, Jennings R, Mibena E, Mabey DCW. Scarring trachoma is associated with polymorphism in the tumor necrosis factor alpha (TNF-α) gene promoter and with elevated TNF-α levels in tear fluid. Infect Immun. 1997;65:1003–6. doi: 10.1128/iai.65.3.1003-1006.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brinkman BM, Zuijdeest D, Kaijzel EL, Breedveld FC, Verweij CL. Relevance of the tumor necrosis factor alpha (TNF alpha) −308 promoter polymorphism in TNF alpha gene regulation. J Inflamm. 1995;46:32–41. [PubMed] [Google Scholar]
- 31.Westendorp RGJ, Langermans JAM, Huizinga TWJ, Elouali AH, Verwij CL, Boomsma DI, Vandenbroucke JP. Genetic influence on cytokine production and fatal meningococcal disease. Lancet. 1997;349:170–3. doi: 10.1016/s0140-6736(96)06413-6. [DOI] [PubMed] [Google Scholar]
- 32.Wilson AG, de Vries N, Pociot F, di Giovine FS, van der Putte LBA, Duff GW. An allelic polymorphism within the human tumor necrosis factor α promoter region is strongly associated with HLA A1, B8, and DR3 alleles. J Exp Med. 1993;177:557–60. doi: 10.1084/jem.177.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy S, McGuire W, Mascie-Taylor CGN, Saha B, Hazra SK, Hill AVS, Kiatkowski D. Tumor necrosis factor promoter polymorphism and susceptibility to lepromatous leprosy. J Infect Dis. 1997;176:530–2. doi: 10.1086/517282. [DOI] [PubMed] [Google Scholar]