

Abstract
T cell lymphopenia in the peripheral blood lymphocytes (PBL) of patients with AT is mainly caused by a decrease of naive CD45RA+/CD4+ cells followed by a predominance of memory CD45RO+ lymphocytes. To relate these findings to the regulation of programmed cell death, we investigated the activation state and apoptotic level of PBL in 12 patients and healthy controls by flow cytometry. In accordance with previous investigations, the number of naive CD4+/CD45RA+ cells was significantly decreased in patients compared with healthy controls. This disturbed balance of CD45RA and CD45RO was also reflected in higher amounts of activated HLA-DR and CD95 expressing cells, with a concomitant decrease of Bcl-2 protected lymphocytes in the T cell population. With regard to its role in preventing oxidative-induced cell death, we analysed Bcl-2 expression and apoptosis in the presence of oxidative stress. In culture, cells of patients are more susceptible to spontaneous programmed cell death. However, in our stress-inducing system (hypoxanthine/xanthine oxidase system) the number of cells undergoing apoptosis was lower in patients' cell populations compared with controls. In addition, preliminary results suggest that Bcl-2 expression and level of spontaneous apoptosis in patients can be modified by IL-2 and interferon-gamma.
Keywords: ataxia telangiectasia, apoptosis, oxidative stress, hypoxanthine/xanthine oxidase system
INTRODUCTION
The syndrome of AT is caused by a single autosomal recessive gene defect on 11q22-23 and includes a variety of apparently unrelated findings such as progressive cerebellar ataxia, telangiectasia, thymic dystrophia with immunodeficiency, chromosomal instability, radiosensitivity, and cancer susceptibility [1–3].
Decline of peripheral lymphocytes in the T cell population is one of the main phenomena of the immunological aspect of the disease. The diminished level of circulating T lymphocytes is mainly due to the absence of CD4+/CD45RA+ cells, which leads to an imbalance between naive and memory cells in the lymphocyte population [4]. This is supported by the diminished functionality of the T cells, including reduced proliferation to mitogens and impaired production of IL-2 and interferon-gamma (IFN-γ) [5,6]. Increased spontaneous apoptosis had been observed in vitro in AT lymphocytes derived from Atm-deficient mice [7,8]; in analogy patients with AT also show an increase in spontaneous apoptosis in lymphoblasts and lymphocytes compared with healthy controls [9].
A defect in the ‘cell surveillance network’ has been proposed to explain the chronic cell loss in the immune system and also in the cerebellum in AT, suggesting that the damaged ataxia telangiectasia-mutated (ATM)-gene product leads to a fault in the cell cycle control and DNA repair [10]. This defect in repairing spontaneous and induced DNA damage leads to increased genomic instability, increased cancer predisposition and an inappropriate regulation of the apoptosis program in AT cells. Features of this dysregulation are reflected in high sensitivity to radiation and radiomimetic drugs, while response is intact in AT cells treated with UV or topoisomerase inhibitors [11–13]. One hypothesis to explain the sensitivity of AT cells to apoptosis is the inability to counteract the effects of reactive oxygen species (ROS) [14]. In high concentrations, these molecules are responsible for DNA damage and the induction of apoptosis [15].
A molecule playing an important role in preventing programmed cell death in an anti-oxidant pathway is the proto-oncogen product Bcl-2 [16,17]. Recently, it has been shown that over-expression of Bcl-2 blocks apoptosis after oxidative damage [18]. Expression of Bcl-2 is down-regulated in T cells after conversation from CD45RA to the CD45RO phenotype in vitro [19]. The elevated apoptosis in AT cells might be due to a decrease in Bcl-2 protection.
In this context, we characterized the CD4+ cell population in AT patients by flow cytometry to analyse their state of activation and their anti-apoptotic level. For this purpose, we studied the expression of HLA-DR, CD95 (Fas/APO-1) and Bcl-2 in relation to the ratio of naive (CD45RA+) and memory (CD45RO+) cells in the AT lymphocyte population. As expected, the expression of these molecules was altered in patients with AT [20,21].
In addition, we investigated spontaneous apoptosis and the influence of oxidative stress on AT lymphocytes in cell culture using an oxygen radical producing system (hypoxanthine/xanthine oxidase system) [22–24]. Our data demonstrate that AT patients are more susceptible to spontaneous apoptosis, but are more resistant to oxygen radicals. Given the high level of spontaneous apoptosis, we investigated whether this could be modified by addition of cytokines. In preliminary experiments we found that IL-2 and IFN-γ were effective in modifying Bcl-2 expression and spontaneous apoptosis in AT patients.
SUBJECTS AND METHODS
Subjects
We studied 12 patients with AT checked regularly at the Frankfurt University Hospital, aged 5–24 years, median 14 years, sex distribution female:male 6:6. AT was diagnosed clinically and by means of increased α-fetoprotein in all patients. Controls for lymphocyte phenotyping were matched for age, and the study of spontaneous and induced apoptosis was carried out with healthy controls.
Lymphocyte phenotyping
Whole blood samples containing EDTA were stained with the following MoAbs directly labelled with FITC or PE: UCHT-1 (CD3), B9.11 (CD8), J4.119 (CD19), ALB11 (CD45RA), UCHL-1 (CD45RO) from Coulter-Immunotech (Marseilles, France), RPA-T4 (CD4), 3G8 (CD16), DX2 (CD95) from PharMingen (San Diego, CA), MY31 (CD56), L-243 (HLA-DR) from Becton Dickinson (San Jose, CA), and C124 (Bcl-2) from Dako Diagnostics (Glostrup, Denmark). After staining, erythrocytes were lysed (FACS-lysing solution; Becton Dickinson), washed with PBS and fixed with 1% paraformaldehyde. For intracellular Bcl-2 staining, cells were fixed and permeabilized with solution A and B (Fix & Perm; An der Grub, Vienna, Austria), Bcl-2 was added to solution B. In each case cells were incubated for 15 min at room temperature.
Samples were analysed by a flow cytometer (FACScan; Becton Dickinson) equipped with an air-cooled 488-nm argon-ion laser. On each sample, 10 000 events were collected and data analysis was performed with Lysis II software (Becton Dickinson).
Hypoxanthine/xanthine oxidase system
Peripheral blood mononuclear cells (PBMC) were separated from heparinized blood by density gradient centrifugation on Ficoll–Hypaque and washed three times with RPMI 1640. Cells were cultured at 37°C, 5% CO2 in RPMI 1640 supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin, 1% HEPES, 2% glutamine and 0.2% gentamycin at a concentration of 1 × 106 cells/ml. For induction of in vitro apoptosis, PBMC were incubated in the presence of hypoxanthine (HX) and xanthine oxidase (XO). As shown in Fig. 2, we used the human lymphoblastoid Jurkat cell line to equilibrate the oxidative stress system. As oxygen radical scavengers we used superoxide dismutase (SOD) (100–10 000 U/ml) and catalase (CAT) (10–1000 U/ml) (Boehringer, Mannheim, Germany).
Fig. 2.
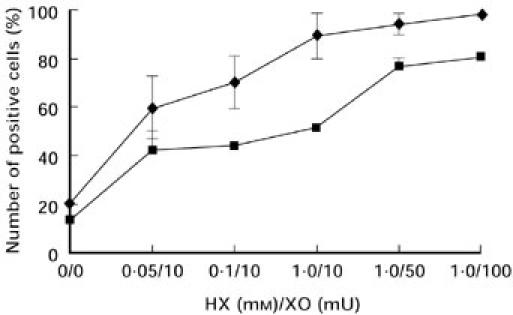
Evaluation of the hypoxanthine/xanthine oxidase (HX/XO) system for the induction of oxidative stress in the lymphoblastoid Jurkat cell line. Stimulation of apoptosis was prepared by various doses of HX and XO after 24 h stimulation and detected by 7-aminoactinomycin D (7AAD; ♦) and propidium iodide (PI; ▪) incorporation. Values represent the means ± s.d. of three separate experiments.
In order to modulate spontaneous apoptosis, IL-2 (Biotest Pharma, Dreieich, Germany) 100 U/ml or IFN-γ (Genzyme, Rüsselsheim, Germany) 250 μg/ml were added to the culture.
Detection of apoptosis
Flow cytometry was used to measure the percentage of apoptosis after 7-aminoactinomycin D (7AAD) staining [25,26]. Briefly, cells were incubated with 20 μg/ml of 7AAD (in PBS/Azide) for 20 min at 4°C in the dark, afterwards cells were analysed within 30 min in their staining solution. For staining cells with hypotonic citrate solution propidium iodide (PI), lymphocytes were washed once with PBS, then 250 ml staining solution (50 μg of PI in 0.1% sodium citrate plus Triton-X; Sigma Aldrich, Deisenhofen, Germany) were added [27]. Cells were kept overnight at 4°C before analysis.
Statistical analysis
Results were expressed as mean ± s.d. and analysed using the Mann–Whitney U-test. P < 0.05 was regarded as significant.
RESULTS
Lymphocyte phenotyping
In accordance with previous investigations, the analysis of AT patients' lymphocyte distribution in peripheral blood (PB) revealed a significant absolute and relative reduction of CD3+ and CD19+ cells (Table 1). T cell lymphopenia was found in all patients, but B cells were reduced in just 75% of the patients. The decrease in T cells, particularly in the CD4 population, was caused by a deficiency of naive CD45RA+ cells (P < 0.001). The absolute number of CD45RO+ cells was not significantly higher compared with healthy controls. However, the reduction of naive cells led to a relative predominance of activated cells in AT cell populations (P < 0.001). These findings were supported by a significant up-regulation of HLA-DR-expressing T cells in relative cell numbers (P < 0.001). Furthermore, we found a relative increase of natural killer (NK) cells in the PB of AT patients (P < 0.001).
Table 1.
Relative and total numbers of lymphocytes in the peripheral blood of AT patients and controls

Results are shown as mean ± s.d. *P < 0.001; **P < 0.005; ***P < 0.01; ****P < 0.05.
As expected, the apoptosis-related cell surface molecule CD95 (Fas/APO-1) was significantly increased in the CD4+ cell population (P < 0.001). The numbers of CD95+ cells correlated well with the amount of memory lymphocytes. Absolute as well as relative numbers of Bcl-2-protected cells were reduced in the CD4+ population (P < 0.05, P < 0.01). The reduction of Bcl-2 expression was mainly seen in CD45RO+ lymphocytes (a typical dot plot analysis is displayed in Fig. 1).
Fig. 1.
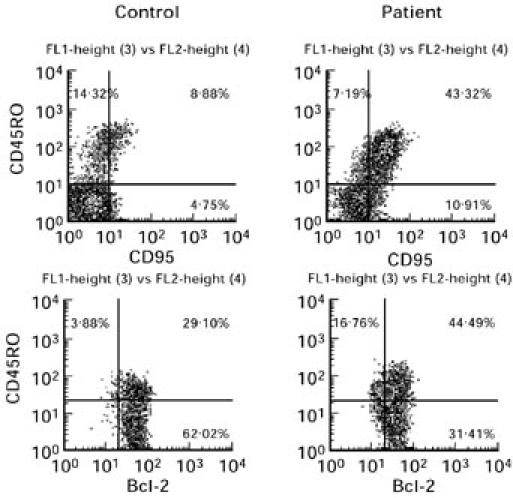
Dot plots from a single representative flow cytometric analysis of CD95/CD45RO (upper panels) and Bcl-2/CD45RO (lower panels) double-stained lymphocytes of AT patients compared with a healthy control.
The hypoxanthine/xanthine oxidase system
Standardizing the HX/XO system, we used the lymphoblastoid Jurkat cell line stimulated with various doses of HX and XO for 24 h. To examine spontaneous and oxidative stress-induced programmed cell death in vitro, membrane permeabilization was detected by 7AAD [26]. We distinguished 7AAD− and 7AAD+ (7AADdim/7AADbright) cells for the differentiation of live and apoptotic cells (early/late apoptotic, dead cells, respectively). As a second method to measure apoptosis, we used PI according to Nicolletti et al. [27]. The detection of both 7AAD as well as PI staining indicates an increase in apoptotic cells after treatment with raised concentrations of oxidative stress (Fig. 2). The staining of hypoploid DNA after oxidative stimulation by PI resulted in a lower rate of apoptosis than 7AAD staining. This reflects the differences of the staining methods as shown by Schmidt et al. [25]. In order to detect all kinds of programmed cell death, including early and late apoptosis, we used the 7AAD staining method for further experiments. Apoptosis induced by the HX/XO system was prevented by extracellular CAT but not by SOD (Fig. 3). These results indicate that hydrogen peroxide rather than the superoxide radical is responsible for oxidative stress-induced apoptosis. The effect of CAT and SOD was analysed in a time kinetic over 144 h (at 24, 48, 72 and 144 h). There was no difference in CAT and SOD action over the whole period (Fig. 4).
Fig. 3.
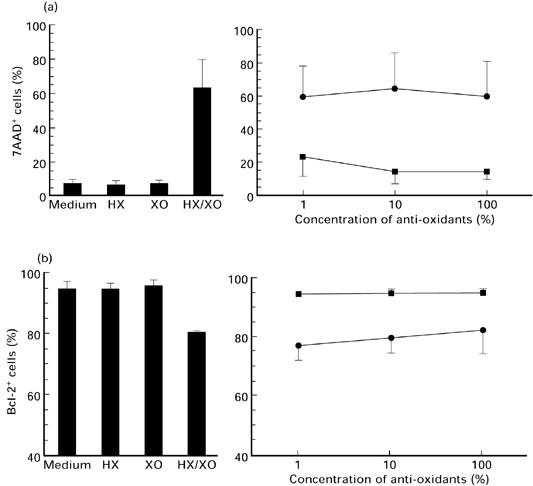
Lymphocytes of healthy individuals treated with hypoxanthine (HX)/xanthine oxidase (XO) (1.0 mm/10 mU/ml),HX and XO alone (left). Dose-dependent effect of catalase (▪) and superoxide dismutase (SOD; •) on cells treated with HX (1.0 mm) and XO (10 mU) for 24 h (right). One hundred percent means 1000 U catalase and 10 000 U SOD. Mean values ± s.d. of (a) 7-aminoactinomycin D (7AAD) incorporation (n = 8) and (b) number of Bcl-2-expressing cells (n = 4) detected by flow cytometry.
Fig. 4.
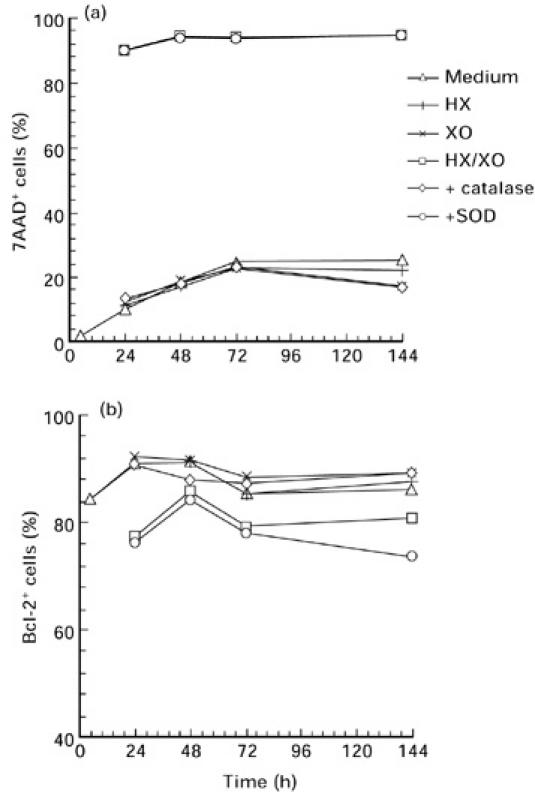
Measurement of (a) 7-aminoactinomycin D (7AAD) incorporation and (b) number of Bcl-2-expressing cells from four healthy individuals by flow cytometry. Lymphocytes were cultured in the presence of hypoxanthine (HX)/xanthine oxidase (XO) (1.0 mm/10 mU/ml), HX and XO alone over a period of 144 h. With regard to the inhibitory effect of the radical scavengers catalase (100 U/ml) or superoxide dismutase (SOD) (1000 U/ml) were added to cell cultures treated with the HX/XO system. Four healthy controls with a strong sensitivity to oxidative stress were examined. Results are shown as mean values.
Spontaneous and oxidative stress-induced apoptosis in AT lymphocytes
We detected spontaneous and oxidative stress-induced apoptosis in AT lymphocytes in medium and after 24-h treatment with 1 mm HX and 10 mU XO. These concentrations were chosen after a series of various stimulations of PBMC (data not shown). Our data of unstimulated apoptosis confirm the results of earlier investigations: AT cells exhibit a higher rate of spontaneous apoptosis (P < 0.05). In contrast, oxidative stress leads to a lower rate of apoptotic cells in AT patients compared with controls (Fig. 5). No differences were observed after phytohaemagglutinin (PHA) stimulation. Simultaneously, the number of Bcl-2-protected cells was lower in AT patients compared with controls in all experiments (Fig. 5).
Fig. 5.
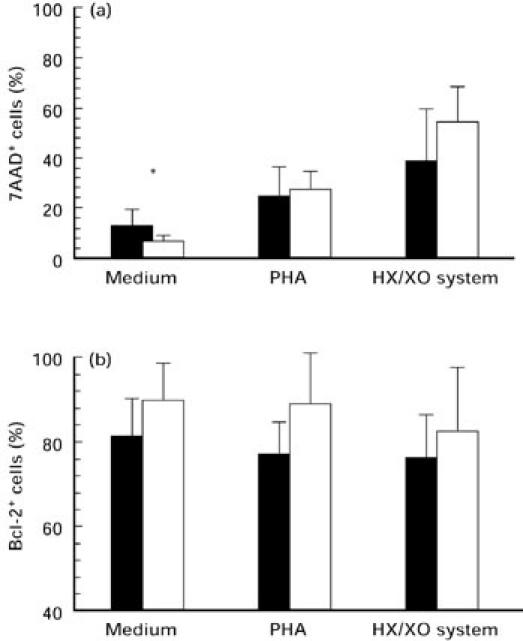
Apoptotic measurement after 24-h stimulation with medium alone, phytohaemagglutinin (PHA) 1 μg/ml and hypoxanthine (HX) 1 mm/xanthine oxidase (XO) 10 mU of AT lymphocytes (▪) in comparison with healthy controls (□). Mean values ± s.d. of the (a) 7-aminoactinomycin D (7AAD) incorporation and (b) the number of Bcl-2-expressing cells were detected by flow cytometry; *P < 0.05.
Next, we investigated the influence of oxidative stress over a period of 144 h. Both apoptotic and Bcl-2-protected lymphocytes were detected after isolation of PBMC and after 24, 48, 72 and 144 h. At all times patients' lymphocytes displayed a higher rate of apoptosis cultured in medium alone, whereas AT cells were more resistant to oxidative stress compared with controls (Fig. 6). Analogous to 7AAD experiments mentioned above, we analysed the effect of CAT and SOD on the number of Bcl-2-expressing cells. CAT but not SOD prevented the down-regulation of Bcl-2+ cells (Fig. 3). These results were also true for the time kinetic experiments (Fig. 4).
Fig. 6.
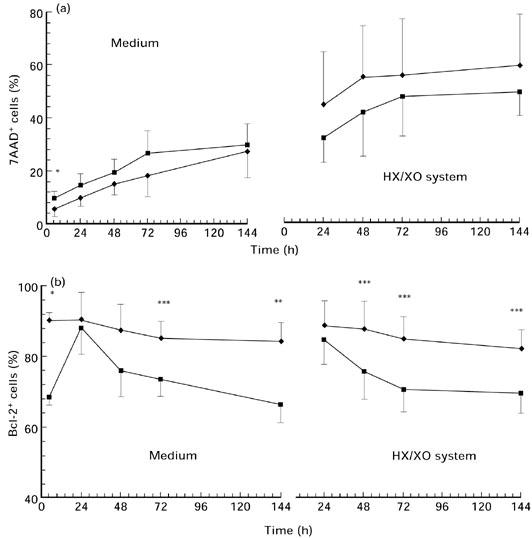
Time kinetics of a 144-h stimulation with hypoxanthine (HX) 1 mm/xanthine oxidase (XO) 10 mU and medium alone. 7-aminoactinomycin D (7AAD) incorporation (a) and Bcl-2 expression (b) were detected after stimulation of AT lymphocytes (▪) and control cells (♦) by flow cytometry. Values are shown as mean ± s.d.; *P < 0.001; **P < 0.01; ***P < 0.05.
In accordance with phenotyping experiments shown in Table 1, the relative amount of Bcl-2-protected cells was significantly lower in the AT cell population than in controls at all times, with an exception at 24 h. At 24 h a sharp rise of Bcl-2+ cells was recorded, whereas control cells exhibited a constant number of Bcl-2-expressing lymphocytes during culture (Fig. 6). No significant differences could be found when cells were stimulated by the HX/XO system.
Given the high level of spontaneous apoptosis, we addressed the question of whether this can be modified by addition of cytokines, e.g. IL-2 and IFN-γ. Since spontaneous apoptosis in patients and controls showed the clearest difference after 72 h culture, we chose this point of time for further experiments. Although only three patients were studied due to lack of material, Table 2 indicates that cytokines may prevent spontaneous apoptosis. At least in two of three patients spontaneous apoptosis was suppressed when cells were co-cultured with IL-2 (100 U/ml) or IFN-γ (250 μg/ml). Moreover, Bcl-2-expressing cells were up-regulated in lymphocytes of AT patients (Table 2).
Table 2.
Modulation of spontaneous apoptosis by addition of IL-2 and IFN-γ

Peripheral blood mononuclear cells (PBMC) from three patients and three controls were cultured in medium alone and in the presence of either IL-2 (100 U/ml) or IFN-γ (250 ng/ml). 7-aminoactinomycin D (7AAD) incorporation and the number of Bcl-2-expressing cells were detected by flow cytometry.
DISCUSSION
A number of studies describe lymphopenia, recurrent infections and cancer in AT. The cellular mechanisms causing lymphopenia, particularly in the CD4+ population, are not fully elucidated, but it is suggested that the decrease of cells is due to a defect in cell cycle control and DNA repair [1–3,10].
Our investigations confirm earlier findings indicating that the lack of lymphocytes is predominantly located in the CD45RA+/CD4+ population, accompanied by a relative increase of activated memory cells [4]. Dysplastic changes of the thymus and a defect in the interconversion of naive and memory lymphocytes in the CD4+ population are discussed as possible explanations for the relative predominance of the low molecular weight CD45 isoform [6,28]. Since B cell numbers and the capacity for antibody production are reduced in AT, the increase of CD45RO+ cells possibly represents a further component in providing help for antigen-induced immune response [29].
The predominance of activated memory cells is accompanied by a significant increase of HLA-DR- and CD95-expressing lymphocytes, reflecting the activation level of these cells. These results have two features in common, as suggested by Saxon [20]: lymphocytes in patients with immunodeficiency exhibit a higher level of activation and an altered apoptotic profile. This is in keeping with our data and a recent report which shows a higher rate of spontaneous apoptosis in AT lymphocytes [9].
The higher activation level of lymphocytes in patients with immunodeficiency, especially seen in AT, might be a pathogenic factor in the course of the disease. In this context, the influence of ROS in signal transduction has been described [30,31]. The susceptibility to ROS is supported by the constitutive activation of the transcription factor NF-κB in AT lymphoblasts [32–34]. The excessive formation of ROS leads to DNA damage and to an apoptotic signal to the cell. A probable sensor of ROS levels and oxidative DNA damage in lymphocytes is the ATM gene product which is involved in the co-ordination of cell cycle control [35].
Taking the importance of ROS in the apoptotic process into account, we applied the HX/XO system which primarily generates superoxide anions and hydrogen peroxide to study oxygen-induced apoptosis in AT cells. The HX/XO system provides a mild form of oxidative stress and was observed to induce apoptosis [22,23]. Induction of apoptosis could be blocked by the radical scavenger CAT, whereas SOD had no effect, indicating that hydrogen peroxide rather than the superoxide radical is responsible for oxidative stress-induced apoptosis. Contrasting with earlier findings that AT fibroblasts demonstrate a higher sensitivity to ROS, we showed that patients' lymphocytes were more resistant to oxidative stress-induced apoptosis than controls, although no statistical significance was reached [31,36]. These data are surprising considering the increased spontaneous apoptosis and are difficult to explain. The hypersensitivity to ionizing radiation and radiomimetic drugs on one hand and the resistance to oxygen radicals on the other may indicate a delayed reaction against cell stress-inducing agents, suggesting that both aspects reflect two distinct characteristics of AT. In analogy, this has also been shown by Enns et al., who did not find any association between radiosensitivity of AT fibroblasts and deregulated apoptosis [37].
A crucial molecule in preventing apoptosis is Bcl-2. As described, expression of Bcl-2 is diminished in CD45RO+ T lymphocytes in patients and controls with acute infections, and consequently T lymphocytes are destined to perish by apoptosis [21,38]. We used Bcl-2 as a marker for protection against apoptosis. Recent studies suggest that the protection is provided by an anti-oxidant pathway [16]. Reduced Bcl-2 expression in AT cells could be demonstrated, whereby the decrease was mainly located in the CD45RO+ population. Similar to the results of lymphocyte phenotyping in whole blood, the level of Bcl-2-expressing cells was significantly decreased in culture. However, only slight reduction of Bcl-2+ cells was found after stimulation with PHA and with the HX/XO system.
An interesting area for further studies is the possibility of using cytokines for modulating the apoptotic state in patients with AT. Since it was previously shown that IL-2 and IFN-γ can restore Bcl-2 expression and promote cell survival of activated T cells derived from individuals with viral infections, we used these cytokines to modify apoptosis. Preliminary experiments in three patients show that this was indeed the case. In this regard it is interesting to note that reduced Bcl-2 mRNA and protein levels have been observed in the cerebellum of mutant mice which experience Purkinje cell degeneration [39].
In conclusion, the number of activated cells reflects the high risk of apoptosis in blood-derived lymphocytes of patients with AT. Interestingly, in our culture system patients' cells were more resistant to oxygen agents such as hydrogen peroxide than control lymphocytes. Moreover, high levels of spontaneous apoptosis can be modified by addition of IL-2 and IFN-γ. Further studies are necessary to investigate to what extent the imbalance between life and death of AT cells can be modulated by cytokines.
Acknowledgments
We thank Gabi Gottwald and Tanja Hase for technical assistance.
REFERENCES
- 1.Gatti RA, Boder E, Vinters HV, Sparkes RS, Norman A, Lange K. Ataxia-telangiectasia: an interdisciplinary approach to pathogenesis. Medicine. 1991;70:99–117. [PubMed] [Google Scholar]
- 2.Lavin MF, Shiloh Y. Ataxia-telangiectasia: a multifaceted genetic disorder associated with defective signal transduction. Curr Opin Immunol. 1996;8:459–64. doi: 10.1016/s0952-7915(96)80030-6. [DOI] [PubMed] [Google Scholar]
- 3.Shiloh Y, Rotman G. Ataxia-telangiectasia and the ATM gene: linking neurodegeneration, immunodeficiency, and cancer to cell cycle checkpoints. J Clin Immunol. 1996;16:254–60. doi: 10.1007/BF01541389. [DOI] [PubMed] [Google Scholar]
- 4.Paganelli R, Scala E, Scarselli E, Ortolani C, Carmini D, Aiuti F, Fiorelli M. Selective deficiency of CD4+/CD45RA+ lymphocytes in patients with ataxia telangiectasia. J Clin Immunol. 1992;12:84–91. doi: 10.1007/BF00918137. [DOI] [PubMed] [Google Scholar]
- 5.Paganelli R, Capobianchi MR, Ensoli B, D'offizi GP, Facchini J, Dianzani F, Aiuti F. Evidence that defective gamma interferon production in patients with primary immunodeficiencies is due to intrinsic incompetence of lymphocytes. Clin Exp Immunol. 1988;72:124–9. [PMC free article] [PubMed] [Google Scholar]
- 6.Waldmann TA, Broder S, Goldmann CK, Korsmeyer SJ, Frost K, Korsmeyer SJ, Medici MA. Disorders of B cells and helper T cells in the pathogenesis of the immunoglobulin deficiency of patients with ataxia telangiectasia. J Clin Invest. 1983;71:282–95. doi: 10.1172/JCI110768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barlow C, Hirotsune S, Paylor R, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–71. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 8.Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P. Pleiotrophic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci USA. 1996;93:13084–9. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duchaud E, Ridet A, Stoppa-Lyonnet D, Janin N, Moustacchi E, Roselli F. Deregulated apoptosis in ataxia telangiectasia: association with clinical stigmata and radiosensitivity. Cancer Res. 1996;56:1400–4. [PubMed] [Google Scholar]
- 10.Meyn MS. Ataxia-telangiectasia and cellular responses to DNA damage. Cancer Res. 1995;55:5591–6001. [PubMed] [Google Scholar]
- 11.Beamish H, Lavin MF. Radiosensitivity in ataxia-telangiectasia: anomalies in radiation-induced cell cycle delay. Int J Radiat Biol. 1994;65:175–84. doi: 10.1080/09553009414550211. [DOI] [PubMed] [Google Scholar]
- 12.Khanna KK, Lavin MF. Ionizing radiation and UV induction of p53 protein by different pathways in ataxia-telangiectasia cells. Oncogene. 1993;8:3307–12. [PubMed] [Google Scholar]
- 13.Canman CE, Wolff AC, Chen CY, Fornance AJ, Jr, Kastan MB. The p53-dependent G1 cell cycle checkpoint pathway and ataxia-telangiectasia. Cancer Res. 1994;54:5054–8. [PubMed] [Google Scholar]
- 14.Rotman G, Shiloh Y. Ataxia-telangiectasia: is ATM a sensor of oxidative damage and stress? Bioessays. 1997;19:911–7. doi: 10.1002/bies.950191011. [DOI] [PubMed] [Google Scholar]
- 15.Buttke TM, Sandstrom PA. Oxidative stress as a mediator of apoptosis. Immunol Today. 1994;15:7–10. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 16.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–51. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 17.Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–20. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 18.Cherbonnel-Lasserre C, Dosanjh MK. Suppression of apoptosis by overexpression of Bcl-2 or Bcl-xL promotes survival and mutagenesis after oxidative damage. Biochimie. 1997;79:613–7. doi: 10.1016/s0300-9084(97)82011-1. [DOI] [PubMed] [Google Scholar]
- 19.Akbar AN, Salmon M, Savill J, Janossy G. A possible role for Bcl-2 in regulating T-cell memory—a ‘balancing act’ between cell death and survival. Immunol Today. 1993;14:526–32. doi: 10.1016/0167-5699(93)90181-J. [DOI] [PubMed] [Google Scholar]
- 20.Saxon A, Keld B, Diaz-Sanchez D, Guo BC, Sidell N. B cells from a distinct subset of patients with common variable immunodeficiency (CVID) have increased CD95 (Apo-1/fas), diminished CD38 expression, and undergo enhanced apoptosis. Clin Exp Immunol. 1995;102:17–25. doi: 10.1111/j.1365-2249.1995.tb06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamaru Y, Miyawaki T, Iwai K, Tsuji T, Nibu R, Yachie A, Koizumi S, Taniguchi N. Absence of bcl-2 expression by activated CD45RO+ T lymphocytes in acute infectious mononucleosis supporting their susceptibility to programmed cell death. Blood. 1993;82:521–7. [PubMed] [Google Scholar]
- 22.Gansauge S, Gansauge F, Gause H, Poch B, Schoenberg MH, Beger HG. The induction of apoptosis in proliferating human fibroblasts by oxygen radicals is associated with a p53- and p21WAFCIP1 induction. FEBS Letters. 1997;404:6–10. doi: 10.1016/s0014-5793(97)00059-8. [DOI] [PubMed] [Google Scholar]
- 23.Dobmeyer TS, Findhammer S, Dobmeyer JM, et al. Ex vivo induction of apoptosis in lymphocytes is mediated by oxidative stress: role for lymphocyte loss in HIV infections. Free Rad Biol Med. 1997;22:775–85. doi: 10.1016/s0891-5849(96)00403-0. [DOI] [PubMed] [Google Scholar]
- 24.Marini M, Frabetti F, Musiani D, Franceschi C. Oxygen radicals induce stress proteins and tolerance to oxidative stress in human lymphocytes. Int J Radiat Biol. 1996;70:337–50. doi: 10.1080/095530096145076. [DOI] [PubMed] [Google Scholar]
- 25.Schmid I, Uittenbogaart CH, Keld B, Giorgi JV. A rapid method for measuring apoptosis and dual-colour immunofluorescence by single laser flow cytometry. J Immunol Methods. 1994;170:145–57. doi: 10.1016/0022-1759(94)90390-5. [DOI] [PubMed] [Google Scholar]
- 26.Philpott NJ, Turner AJ, Scopes J, Westby M, Marsh JC, Gordon-Smith EC, Dalgleish AG, Gibson FM. The use of 7-amino actinomycin D in identifying apoptosis: simplicity of use and broad spectrum of application compared with other techniques. Blood. 1996;87:2244–51. [PubMed] [Google Scholar]
- 27.Nicoletti I, Migliorati G, Pagliacci MC, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–9. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 28.Bell EB, Sparshott SM. Interconversion of CD45R subsets of CD4 T cells in vivo. Nature. 1990;348:163–6. doi: 10.1038/348163a0. [DOI] [PubMed] [Google Scholar]
- 29.Richards SJ, Scott CS, Cole JC, Gooi HC. Abnormal CD45R expression in patients with common variable immunodeficiency and X-linked agammaglobulinaemia. Br J Haematol. 1992;81:160–6. doi: 10.1111/j.1365-2141.1992.tb08201.x. [DOI] [PubMed] [Google Scholar]
- 30.Yi M, Rosin MP, Anderson CK. Response of fibroblast cultures from ataxia-telangiectasia patients to oxidative stress. Cancer Lett. 1990;54:43–50. doi: 10.1016/0304-3835(90)90089-g. [DOI] [PubMed] [Google Scholar]
- 31.Ward AJ, Olive PL, Burr AH, Rosin MP. Response of fibroblast cultures from ataxia-telangiectasia patients to reactive oxygen species generated during inflammatory reactions. Environ Mol Mutagen. 1994;24:103–11. doi: 10.1002/em.2850240205. [DOI] [PubMed] [Google Scholar]
- 32.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10:709–20. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 33.Jung M, Zhang Y, Lee S, Dritschilo A. Correction of radiation in ataxia telangiectasia cells by a truncated IκB-alpha. Science. 1995;268:1619–1. doi: 10.1126/science.7777860. [DOI] [PubMed] [Google Scholar]
- 34.Jung M, Kondratyev A, Lee SA, Dimtchev A, Dritschilo A. ATM gene product phosphorylates IκB-alpha. Cancer Res. 1997;57:24–7. [PubMed] [Google Scholar]
- 35.Rotman G, Shiloh Y. The ATM gene and protein: possible roles in genome surveillance, checkpoint controls and cellular defence against oxidative stress. Cancer Surv. 1997;29:285–304. [PubMed] [Google Scholar]
- 36.Green MH, Marcovitch AJ, Harcourt SA, Lowe JE, Green IC, Arlett CF. Hypersensitivity of ataxia-telangiectasia fibroblasts to a nitric donor. Free Radic Biol Med. 1997;22:343–7. doi: 10.1016/s0891-5849(96)00336-x. [DOI] [PubMed] [Google Scholar]
- 37.Enns L, Barley RD, Paterson MC, Mirzayans R. Radiosensitivity in ataxia telangiectasia fibroblasts is not associated with deregulated apoptosis. Radiat Res. 1998;150:11–6. [PubMed] [Google Scholar]
- 38.Akbar AN, Borthwick N, Salmon M, et al. The significance of low bcl-2 expression by CD45RO T cells in normal individuals and patients with acute viral infections. The role of apoptosis in T cell memory. J Exp Med. 1993;178:427–38. doi: 10.1084/jem.178.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gillardon F, Baurle J, Wickert H, Grüsser-Cornehls U, Zimmermann M. Differential regulation of bcl-2, bax, c-fos, junB, and krox-24 expression in the cerebellum of Purkinje cell degeneration mutant mice. J Neurosci. 1995;41:708–15. doi: 10.1002/jnr.490410517. * [DOI] [PubMed] [Google Scholar]