

Abstract
ICAM-1 (CD54), the ligand for LFA-1 and Mac-1, is up-regulated during inflammatory reaction on the activated vascular endothelium. To determine its role in intestinal inflammation, we induced acute experimental colitis in mice with a deleted ICAM-1 gene, by feeding them with 3% dextran sodium sulphate (DSS) in drinking water for 7 days. Chronic colitis was elicited by DSS similarly, followed by 2 weeks with water. In the acute phase of inflammation, ICAM-1-deficient mice exhibited a significantly lower mortality rate (5%) than control C57Bl/6J mice (35%). Control animals, but not the ICAM-1-deficient mice, exhibited diarrhoea and rectal bleeding. Histological examination of large-bowel samples evaluated the intensity of inflammatory changes, and type and extent of mucosal lesions. In the acute phase, 33.3% of samples from ICAM-1-deficient mice exhibited mucosal defects (flat and fissural ulcers), predominantly mild to moderate inflammatory infiltrate within the lamina propria mucosae and lower grades of mucosal lesions. Much stronger inflammatory changes were present in control animals, flat ulcers (sometimes multiple) and fissural ulcers being observed in 62.5% of samples. Mucosal inflammatory infiltrate was moderate to severe, typically with higher grades of mucosal lesions. In chronic colitis, smaller inflammatory changes were found in the large bowel. The two mouse strains differed, the chronic colitis being accompanied by an increased serum level of anti-epithelial IgA autoantibodies in C57Bl/6 control mice but not in ICAM-1-deficient mice. These findings provide direct evidence of the participation of ICAM-1 molecule in the development of experimentally induced intestinal inflammation.
Keywords: intercellular adhesion molecule-1, intestinal inflammation, ulcerative colitis adhesion molecules, gene knock-out
INTRODUCTION
Intestinal inflammation is characterized by the accumulation of blood-borne polymorphonuclear leucocytes, monocyte/macrophages, and naive and memory T cells within submucosal intestinal epithelium [1]. Emigration of leucocytes to the site of inflammation has been described to rely on an initial contact of circulating leucocytes with the activated endothelium which leads, via the interactions of sequentially acting receptor/counter-receptor pairs, to the firm attachment of the rolling cells to the endothelium (for review see [2]). These steps include several groups of molecules, namely selectins (P-, E- and L-selectin) and their ligands (PSGL-1, ESL-1), chemoattractants and their receptors, α4-integrin (VLA-4, CD49d), β2-integrins LFA-1 and Mac-1, and immunoglobulin superfamily members such as ICAM-1 (CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106). Although interactions of these individual players represent a possible mechanism in the development of acute or chronic colitis, the particular contribution of these molecules in the initiation and progression of inflammatory bowel disease (IBD) remains to be determined. The importance of these molecules in inflammatory conditions of the gastrointestinal tract is suggested by a recent paper [3], in which an inhibitor of β2-integrin expression (NPC 15669) attenuated acetic acid-induced colitis in rats.
ICAM-1 is a cell adhesion molecule which is up-regulated on activated vascular endothelium upon stimulation via IL-1, IL-17, tumour necrosis factor-alpha (TNF-α), interferon-gamma (IFN-γ), or lipopolysaccharide (LPS) [4–8], and it is also expressed on lymphocytes, monocytes, and epithelial cells [9]. This molecule has been described to be a natural ligand for LFA-1 and Mac-1 integrins. ICAM-1 is a type I membrane glycoprotein consisting of five immunoglobulin-like extracellular domains, a transmembrane-spanning domain, and a short cytoplasmic tail [7,10,11]. The gene for ICAM-1 is composed of seven exons. Exon 1 encodes the signal sequence, exons 2–6 encode extracellular immunoglobulin domains 1–5, respectively, and exon 7 encodes the transmembrane-spanning domain plus cytoplasmic tail [12,13]. Two groups developed mouse strains with targeted mutations in the ICAM-1 locus through the disruption of exon 5 [14], or exon 4 [15]. Further studies with ICAM-1-deficient mice have shown that these mice are resistant to septic shock [15], protected against ischaemic renal injury [16], and cerebral ischaemia reperfusion injury [17]. A recent study indicates that ICAM-1 plays a major role in the pathogenesis of glomerulonephritis and participates in vascular injury in Faslpr mice [18].
The present study was designed to seek definitive evidence for the role of ICAM-1 in acute and chronic intestinal inflammation. To this goal, we induced colitis in mice with disrupted ICAM-1 gene, and in control C57Bl/6J mice, by an oral administration of dextran sodium sulphate (DSS) [19]. We performed detailed analysis of the severity of clinical symptoms of colitis and evaluated the intensity and extent of the inflammatory changes in the large bowel using a histochemical technique. The study was completed by the determination of epithelial autoantibody production in this experimental model of colitis.
MATERIALS AND METHODS
Animals
ICAM-1-deficient mouse strain C57Bl/6J-ICAM-1tm1Bay [14] was originally obtained from the Jackson Laboratory (Bar Harbor, ME), and has been propagated in our specific pathogen-free (SPF) breeding colony with a routine testing for ICAM-1 gene mutation using the polymerase chain reaction (PCR) technique. The control mice C57Bl/6J were purchased from Top VELAZ Ltd (Prague, Czech Republic). All mice were female, 3 months old and weighed around 20 g. The animals were purchased as specific pathogen-free, and were kept under standard conditions. During experiments, mice of both strains were kept in the same room.
Induction of colitis
Acute colitis was induced in wild-type or ICAM-1-deficient mice by addition of 3% DSS (w/v 40 kD) into drinking water for 7 days [19]. To induce chronic colitis DSS treatment was followed by 2 weeks of water [19]. Control mice received only water.
Clinical evaluation
In the course of DSS treatment, clinical symptoms and mortality were checked daily. Animals were evaluated for stool consistency (presence of diarrhoea), rectal prolapse, presence of rectal bleeding and lethargy.
Pathological and histological evaluation of colitis
After the mice were killed (day 7 in acute colitis and day 21 in chronic colitis), blood was taken and the colon was removed. The length of the colon from the ileocaecal junction to the anal verge was measured. For morphological evaluation, the colon was divided into three separate parts (ascendant, transverse, and descendent) and fixed in 4% formalin solution. The samples were randomly taken from each part of the large bowel of both groups of animals. Each segment was submitted for histological processing. All slides were stained with haematoxylin–eosin and serial four to seven sections from each paraffin block sample were examined under the light microscope. Histological observation of the samples was orientated predominantly to the estimation of the grade of mucosal lesions and the presence of mucosal defects (ulcers) and their characterization. The intensity of mucosal inflammatory infiltrate, the changes in goblet cells (reduction or hyperplasia) and crypt epithelium were used for more detailed characterization of the inflammatory process. The crypt scoring was based on the method used by Cooper et al. [20]: grade 0, intact crypt; grade 1, loss of the basal one-third of the crypt; grade 2, loss of the basal two-thirds of the crypt; grade 3, loss of the entire crypt. We evaluated also the presence of flat, fissural or aphtoid ulcers. Flat ulcers were defined as large mucosal defects, the fissural ulcers are narrow deep mucosal defects, and aphtoid ulcers are mucosal defects that appear above the intestinal lymphatic tissue. The inflammatory infiltrate in the mucosa was scored semiquantitatively as minimal, mild, moderate and severe. The scoring observations were performed blindly, in ignorance of ICAM-1 status; the histological slides were marked with numbers only.
ELISA for anti-epithelial antibodies
In order to study the reactivity of serum antibodies from mice with chronic colitis we used isolated intestinal epithelial cells. For the preparation of these cells, female Wistar rats aged 2–3 months from the Institute of Physiology, ASCR, Prague, were used. Intestinal cells were isolated essentially by the method described before [21]. Suspensions of isolated cells were centrifuged at 4°C and washed twice in PBS. Microtitre plates were coated with isolated epithelial cells (5 × 106 cells/10 ml, 50 μl per well), incubated overnight at 4°C, and sediment cells were fixed for 10 min at room temperature with 0.5% glutaraldehyde. After several washings with PBS, the diluted sera were applied and ELISA was performed as described previously with minor modifications [22]. Optimal dilutions of sheep peroxidase-conjugated anti-mouse IgA and IgG were used (The Binding Site, Birmingham, UK). Samples of pooled sera from BALB/c mice were used as a reference serum. The results are expressed as percentage of internal reference serum values.
Statistical analysis
The results are expressed as mean ± s.e.m. Statistical analysis was performed using Student's t-test for unpaired data (two-tail) and P < 0.05 was considered statistically significant.
RESULTS
Lower mortality and missing clinical symptoms of colitis in DSS-treated ICAM-1-deficient mice
An oral administration of DSS was used to evaluate the clinical and pathological changes in ICAM-1-deficient and control (+/+) mice in acute and chronic intestinal inflammation. The clinical symptoms of intestinal inflammation, i.e. diarrhoea, rectal bleeding and rectal prolapse, were observed on day 6 of DSS feeding. Clinical findings for acute colitis presented in Table 1 show that the symptoms of colitis were found only in wild-type animals. Diarrhoea, rectal prolapse and lethargy were not detected in ICAM-1-deficient mice. The mortality in +/+ mice (35%, n = 23) was substantially higher than in ICAM-1-deficient mice (5%, n = 22).
Table 1.
Clinical evaluation—acute colitis

*Indicates the percentage of animals with the symptom.
x/n, Number of affected animals to the total number of analysed animals.
Histological findings indicate that ICAM-1-deficient mice are protected from severe forms of intestinal inflammation
Acute colitis
Post-mortem examination and histological evaluation were performed to test whether the deficiency of ICAM-1 molecules influences the development of colitis. In comparison with mice receiving drinking water, oral administration of 3% DSS induced shortening of the colon; the colon length was decreased by 36% in control mice and by only 26% in ICAM-1-deficient mice. This difference was statistically significant (P = 0.0278). The results of histological examination of the colon are presented in Table 2. In C57Bl/6 mice, shortening of crypts (grade 1, 2 or 3 lesions) in 23 of 24 examined samples (95.9%) was found. Single ulcers were found in 15 samples (62.5%), while multiple ulcers were present in five samples (20.9%). Flat ulcers, defined as large mucosal defects, represented the most frequent type of mucosal defect (25.0%) (Fig. 1A). Fissural, narrow, deep ulcers (Fig. 1B) and aphtoid ulcers above the lymphoid nodules located in the mucosa of the large bowel were observed in 12.5% and 4.1% of samples, respectively. Changes of goblet cells (reduction of hyperplasia) were found in 37.5% of samples. The thinning of crypt epithelium and the presence of bleeding were found in 62.5% and 58.3% of samples, respectively. Submucosal oedema was found in 61.6% of samples, and in 45.0% of samples the oedema was severe. Microscopic evaluation showed also an inflammatory infiltrate of the mixed type (lymphocytes and polymorphonuclear leucocytes) in the lamina propria. Quantitatively, the infiltrate was predominantly moderate (50.0%) and severe (37.5%). Transmural inflammation was observed in 79.1% of samples. All morphological changes were not equally distributed and the damage to the mucosa as well as the extent of inflammation within the large-bowel wall were not uniform.
Table 2.
Histological evaluation of acute dextran sodium sulphate (DSS)-induced colitis

Control C57Bl/6J mice, n = 24 samples; ICAM-1-deficient mice, n = 18 samples.
*Grade 1 to grade 3, Grades of crypt shortening (for description see Materials and Methods).
Fig. 1.
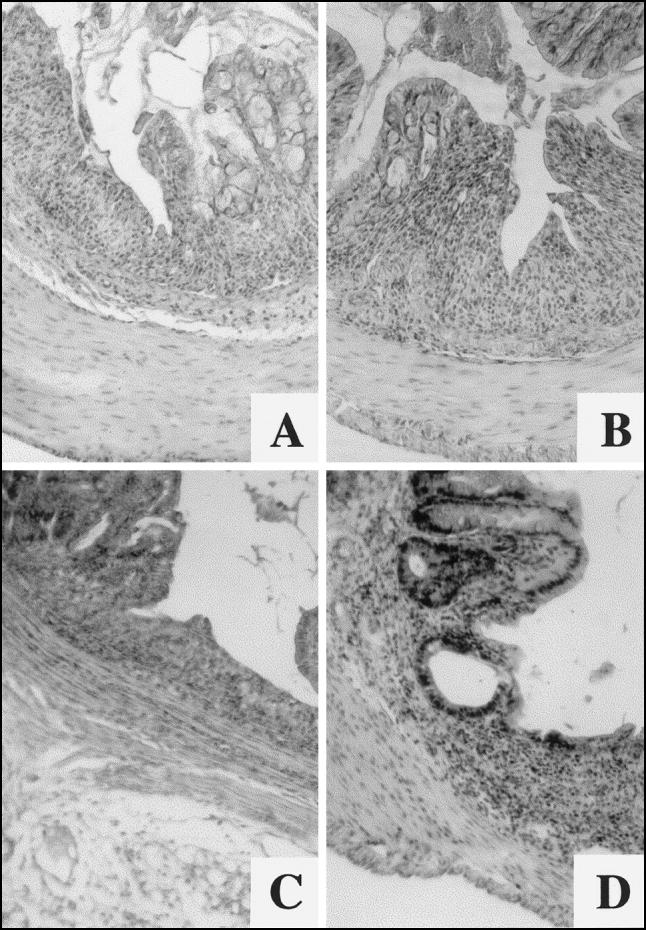
Histopathological manifestations of acute colitis. C57Bl/6 control mice: (A) flat ulcer and hypersecretion of mucin in adjacent colonic mucosa; (B) narrow fissural ulcer. ICAM-1-deficient mice: (C) flat ulcer; (D) grade 3 lesion with cystic dilatation of the crypt and goblet cell reduction. (Haematoxylin and eosin, mag. × 63.)
In comparison with wild-type mice, microscopic changes in ICAM-1-deficient animals were much less extensive (Table 2). In these animals the ulcers were found only in six samples (33.3%). The majority of mucosal defects revealed narrow fissural appearance and no aphtoid or multiple ulcers were found. Flat ulcers were observed only exceptionally (Fig. 1C). In no sample was bleeding observed. Mucosal lesions were found in 15 of 18 samples (83.4%), with the predominance of lower grade lesions. Mild submucosal oedema was present in 11 of 18 samples (61.1%). In 13 of 18 samples (72.3%), reduction of goblet cells was found but only one of 18 samples showed focal hyperplasia of goblet cells. Microscopic examination discovered also an inflammatory infiltration of the mixed type (lymphocytes and granulocytes) within the lamina propria, of predominantly moderate intensity.
Chronic colitis
The histological appearance of the chronic phase of DSS-induced colitis was characterized by less prominent changes in large-bowel mucosa, as well as in submucosa, compared with the acute phase. All lesions found in chronic colitis were focal. Significant differences were found between wild-type mice and ICAM-1-deficient mice (Table 3). While mucosal defects (ulcers) were found in nine samples of 21 (42.9%) wild-type animals, one ulcer only was present in 24 samples (4.0%) of ICAM-1-deficient animals. Flat ulcers represented the most frequent type of mucosal defects in samples obtained from the wild-type strain.
Table 3.
Histological evaluation of chronic dextran sodium sulphate (DSS)-induced colitis
Control C57Bl/6J mice, n = 21 samples; ICAM-1-deficient mice, n = 24 samples.
*Grade 1 to grade 3, Grades of crypt shortening (for description see Materials and Methods).
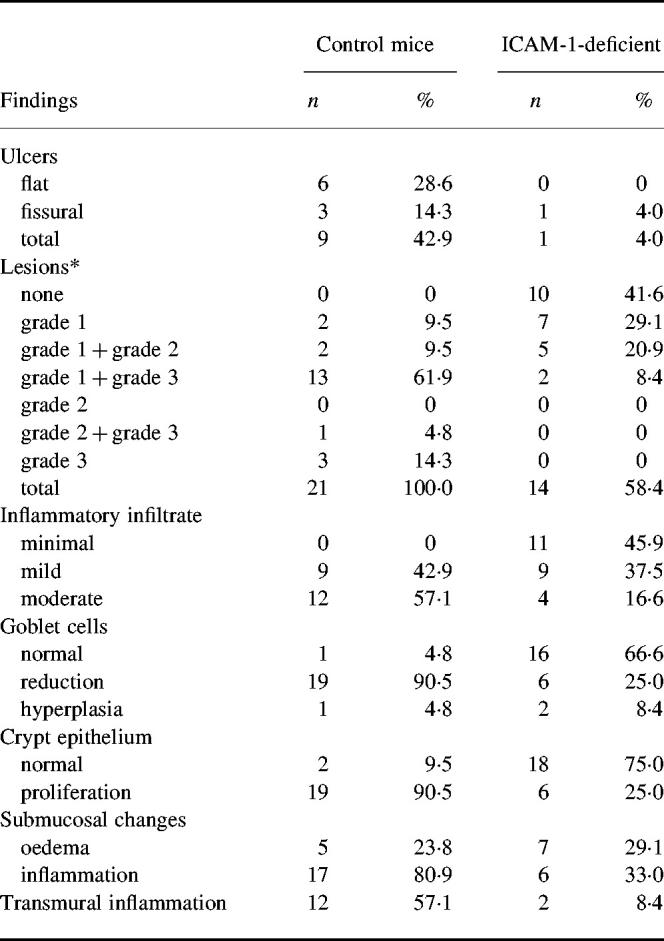
Shortening of crypts (grade 1 to grade 3 lesions) was observed in all samples from wild strain (100.0%), but only in 14 of 24 samples (58.4%) were such changes found in ICAM-1-deficient animals. Larger grade 3 and focal grade 1 to grade 3 lesions together in one sample with prominent goblet cell reduction in adjacent mucosa predominated in the wild-type animals (Fig. 2A,B). Focal grade 1 to grade 2 lesions represented the most frequent alterations of large-bowel mucosa in ICAM-1-deficient animals, while focal grade 1 to grade 3 lesions were rather exceptional in this strain of mice. Goblet cell reduction and proliferation of crypt epithelium were found in the vicinity of such lesions (Fig. 2C,D).
Fig. 2.
Histopathological manifestations of chronic phase of colitis. C57Bl/6 control mice: (A) large grade 3 lesion, probably re-epithelized flat ulcer; (B) grade 2 lesion. ICAM-1-deficient mice: (C) grade 1–3 lesion. (D) Goblet cell reduction and proliferation of crypt epithelium are present in the vicinity of the lesion. (Haematoxylin and eosin, mag. × 63.)
In both strains of animals, signs of epithelial regeneration were found, especially in the vicinity of the ulcers.
Low production of anti-epithelial IgA autoantibodies in DSS-treated ICAM-1-deficient mice
Control and ICAM-1-deficient mice with DSS-induced chronic colitis were tested for the presence of serum anti-epithelial cell antibodies. Our results show that the levels of IgA autoantibodies in wild-type mice were statistically significantly higher than in water drinking mice, whereas only low levels of these IgA autoantibodies were found in DSS-treated ICAM-1-deficient mice, the difference between wild-type and ICAM-1-deficient mice after DSS feeding being statistically significant (P = 0.0006) (Fig. 3). No statistically significant differences were found in levels of IgG antibodies (Fig. 3).
Fig. 3.
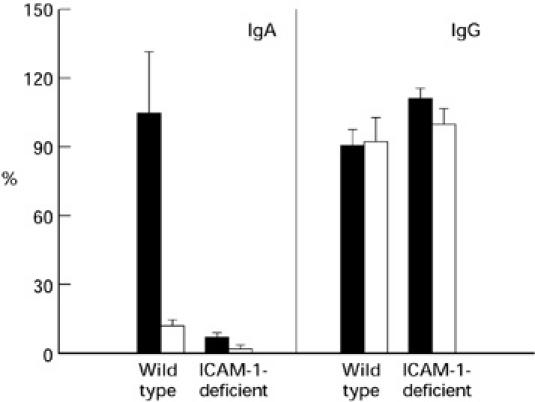
Presence of anti-epithelial antibodies determined by ELISA in sera of ICAM-1-deficient and control mice with chronic colitis (seven mice were used per group). Results are expressed as a percentage of the reactivity of reference serum. Values are indicated as the mean ± s.e.m. of each group. Statistically significant differences were found in the levels of IgA antibodies (at P ≤ 0.001) between dextran sodium sulphate (DSS)-treated ICAM-1-deficient and wild-type control mice and between DSS-induced and untreated mice. ▪, DSS; □, water.
DISCUSSION
DSS given orally to mice and other rodents has been shown to produce intestinal inflammation [19,23]. We used the model of DSS-induced colitis in ICAM-1-deficient mice to address the question of the participation of the ICAM-1 molecule in the development of intestinal inflammation. The results of our experiments indicate that ICAM-1 is one of the essential molecules involved in an effector mechanism leading to severe intestinal damage. Our findings are in agreement with previous data where the role of ICAM-1 in development of an experimental colitis was suggested by amelioration of inflammation after anti-ICAM-1 antibody administration to rats [24] and by anti-sense oligonucleotide ISIS 3082 treatment in mice [25].
The pathogenic mechanisms of DSS-induced colitis are not fully understood. However, recently it has been proposed that intestinal inflammation is related to DSS-mediated toxic damage of colonic crypt epithelium and increased mucosal permeability. Besides this, enhanced colonic endothelial ICAM-1 expression followed by cell infiltration are early events in the inflammatory cascade [26]. In our study we have shown that ICAM-1 expression is up-regulated in wild-type mice after DSS feeding, but not in ICAM-1 deficient mice [27].
Our finding of different intensity and morphological picture of the inflammatory process in the large bowel in the acute and chronic stages suggests that ICAM-1 plays a role especially in early phases of the inflammatory process and that it does not influence healing mechanisms so intensively.
The participation of the ICAM-1 molecule in the pathogenesis of human IBD was demonstrated by enhanced expression of this molecule on colonic endothelial cells in tissue specimens and by increased levels of the soluble form of ICAM-1 in sera and mucosal specimens of patients with Crohn's disease (CD) and ulcerative colitis (UC) [4,28–31]. Moreover, the possible association of ICAM-1 gene polymorphism with heterogeneous subsets of patients with CD and UC has recently been described [32]. Despite the lack of a closer similarity between the experimental mouse and particular type of human IBD, this, as well as other models with targeted gene mutations, represents a valuable tool for analysing the pathogenic mechanisms of intestinal inflammation. However, it has to be pointed out that human IBD in contrast to DSS-induced colitis is a long-lasting disease mediated by specific components of the immune system.
The presence of various autoantibodies in sera from patients with IBD was repeatedly demonstrated [33]. On the basis of their findings Das and co-workers suggested the co-expression of the epitopes of ICAM-1 molecule and autoantigen mol. wt 40 kD on epithelial cells activated by cytokines [34]. The induction of anti-epithelial cell antibodies by DSS feeding in wild-type mice detected in our study indicates that these autoantibodies appear as a consequence of epithelial cell damage occurring during the inflammatory process. The IgA isotype of these antibodies indicates that they probably originate in mucosal associated lymphatic tissue. A lower production of IgA autoantibodies in ICAM-1-deficient mice would therefore correspond to a lower grade of epithelial damage.
Gene ablation techniques have already shown a qualitatively new approach in an effort to elucidate the significance of various immune factors in the development of human diseases. One of the most important observations arising from the studies with genetically deficient mouse models is the finding that deletion of genes involved in immunoregulatory T cell functions results in the development of a spontaneous inflammation affecting preferentially the gut mucosa: mouse strains with deficiency in TCR α-chain, TCR β-chain, MHC class II, IL-2, IL-10 and G signalling protein subunit (G α2) generally develop a chronic inflammation affecting, to a different extent, various parts of the gut (for review see [35]). These results support the idea that immunoregulatory defects in the mucosal response (probably because of a breakdown of oral tolerance) to microflora antigens are substantially involved in pathogenic mechanisms of chronic intestinal inflammation, and bring new insights into the aetiology and pathogenesis of these important disorders [23,36–38].
Animal models with targetted deletion of genes coding for adhesion molecules have not been used for investigation of their role in the intestinal inflammation. Our effort therefore represents the first step in this direction. Our findings confirm that ICAM-1 participates in the development of severe forms of colitis and suggest that blocking the function of this molecule can represent a pivotal therapeutic intervention in this disease.
Acknowledgments
The authors wish to thank B. Cukrowska and P. Rossmann for help in the experiments and valuable discussions, D. Horáková for technical assistance and P. Dráber for critical reading of the manuscript. This study was supported by grant A7020716 (H.T.-H.) from the Grant Agency of the Academy of Sciences of the Czech Republic, grants 301/97/0234 (P.M.), 306/98/0433 (H.T.-H.) and 310/96/1366 (L.T.) from the Grant Agency of the Czech Republic, grants 3761–3 (H.T.-H.) and 5051-3 (L.T.) from the Internal Grant Agency of the Czech Ministry of Health and grant VS96149 (L.P.) from the Grant Agency of Ministry of Education, Youth and Sport.
REFERENCES
- 1.Burgio VL, Fais S, Boirivant M, Perrone A, Pallone F. Peripheral monocyte and naive T-cell recruitment and activation in Crohn's disease. Gastroenterology. 1995;109:1029–38. doi: 10.1016/0016-5085(95)90560-x. [DOI] [PubMed] [Google Scholar]
- 2.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the mutistep paradigm. Cell. 1994;76:301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 3.Noronha-Blob L, Lowe VC, Mulhauser RO, Burch RM. NPC 15669, an inhibitor of neutrophil recruitment is efficacious in acetic acid-induced colitis in rats. Gastroenterology. 1993;104:1021–9. doi: 10.1016/0016-5085(93)90269-i. [DOI] [PubMed] [Google Scholar]
- 4.Jones SC, Banks RE, Haidar A, et al. Adhesion molecules in inflammatory bowel disease. Gut. 1995;36:724–30. doi: 10.1136/gut.36.5.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pober JS, Gimbrone MA, Lapierre LA, et al. Overlapping patterns of activation of human endothelial cells by interleukin 1, tumor necrosis factor and immune interferon. J Immunol. 1986;137:1893–6. [PubMed] [Google Scholar]
- 6.Yao Z, Painter SL, Fanslow WC, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483–6. [PubMed] [Google Scholar]
- 7.Simmons D, Makgoba MW, Seed B. ICAM-1, an adhesion ligand of LFA-1, is homologous to the neural cell adhesion molecule NCAM. Nature. 1988;331:624–7. doi: 10.1038/331624a0. [DOI] [PubMed] [Google Scholar]
- 8.Dustin ML, Rothlein R, Bahn AD, Dinarello CA, Springer TA. Induction by IL-1 and interferon: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1) J Immunol. 1986;137:245–54. [PubMed] [Google Scholar]
- 9.Bevilacqua MP. Endothelial-leukocyte adhesion molecules. Ann Rev Immunol. 1993;11:767–804. doi: 10.1146/annurev.iy.11.040193.004003. [DOI] [PubMed] [Google Scholar]
- 10.Ballantyne CM, O'brien WE, Beaudet AL. Nucleotide sequence of the cDNA for murine intercellular adhesion molecule-1 (ICAM-1) Nucl Acids Res. 1989;17:5853. doi: 10.1093/nar/17.14.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horley KJ, Carpenito C, Baker B, Takei F. Molecular cloning of murine intercellular adhesion molecule (ICAM-1) EMBO J. 1989;10:2889–96. doi: 10.1002/j.1460-2075.1989.tb08437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voraberger G, Schafer R, Stratowa C. Cloning of the human gene for intercellular adhesion molecule-1 and analysis of its 5′-regulatory region. Induction by cytokines and phorbol ester. J Immunol. 1991;147:2777. [PubMed] [Google Scholar]
- 13.Ballantyne CM, Sligh JE, Dai XY, Beaudet AL. Characterization of the murine ICAM-1 gene. Genomics. 1992;14:1076–80. doi: 10.1016/s0888-7543(05)80132-6. [DOI] [PubMed] [Google Scholar]
- 14.Sligh JE, Ballantyne CM, Rich SS, et al. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule-1. Proc Natl Acad Sci USA. 1993;90:8529–33. doi: 10.1073/pnas.90.18.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu H, Gonzalo JA, Pierre Y, et al. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J Exp Med. 1994;180:95–109. doi: 10.1084/jem.180.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly KJ, Williams WW, Jr, Colvin RB, et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemical renal injury. J Clin Invest. 1996;97:1056–63. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soriano SG, Lipton SA, Wang YF, et al. Intercellular adhesion molecule-1-deficient mice are less susceptible to cerebral ischemia-reperfusion injury. Ann Neurol. 1996;39:618–24. doi: 10.1002/ana.410390511. [DOI] [PubMed] [Google Scholar]
- 18.Bullard DC, King PD, Hicks MJ, Dupont B, Beaudet AL, Elkon KB. Intercellular adhesion molecule-1 deficiency protects MRL/MpJ-FasIpr mice from early lethality. J Immunol. 1997;159:2058–67. [PubMed] [Google Scholar]
- 19.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 20.Cooper HS, Murthy SNS, Shah RS, Sedegran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–49. [PubMed] [Google Scholar]
- 21.Kolínská J, Kraml J, Zákostelecká M, Kadlecová L. Actinomycin D suppressed hydrocortisone-induced changes in distribution and sialylation of brush-border enzymes in jejunal villi and crypts. Biochem Int. 1990;21:581–91. [PubMed] [Google Scholar]
- 22.Tučková L, Tlaskalová-Hogenová H, Farré MA, et al. Molecular mimicry as a possible cause of autoimmune reactions in celiac disease? Antibodies to gliadin cross-react with epitopes on enterocytes. Clin Immunol Immunopathol. 1995;74:170–6. doi: 10.1006/clin.1995.1025. [DOI] [PubMed] [Google Scholar]
- 23.Elson CO, Sartor RB, Tennyson GS, Ridell RH. Experimental models of inflammatory bowel disease. Gastroenterology. 1995;109:1344–67. doi: 10.1016/0016-5085(95)90599-5. [DOI] [PubMed] [Google Scholar]
- 24.Wong PY, Yue G, Yink Miyasaka M, et al. Antibodies to intercellular adhesion molecule-1 ameliorate the inflammatory response in acetic acid-induced inflammatory bowel disease. J Pharmacol Exp Ther. 1995;274:475–80. [PubMed] [Google Scholar]
- 25.Bennett CF, Kornbrust D, Henry S, et al. An ICAM-1 antisense oligonucleotide prevents and reverses dextran sulfate sodium-induced colitis in mice. J Pharmacol Exp Ther. 1997;280:988–1000. [PubMed] [Google Scholar]
- 26.Breider MA, Eppinger M, Gough A. Intercellular adhesion molecule-1 expression in dextran sulphate induced colitis in rats. Vet Pathol. 1997;34:598–604. doi: 10.1177/030098589703400608. [DOI] [PubMed] [Google Scholar]
- 27.Bendjelloul F, Rossmann P, Malý P, Mandys V, Jirkovská M, Prokešová L, Tučková L, Tlaskalová-Hogenová H. The detection of ICAM-1 in experimentally induced colitis of ICAM-1 deficient and wild-type mice: an immunohistochemical study. Submitted. doi: 10.1023/a:1004191825644. [DOI] [PubMed] [Google Scholar]
- 28.Bernstein CN, Sargent M, Gallatin WM. Beta2 integrin/ICAM expression in Crohn's disease. Clin Immunol Immunopathol. 1998;86:147–60. doi: 10.1006/clin.1997.4462. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen OH, Langholz E, Hendel J, Brynskov J. Circulating soluble intercellular adhesion molecule-1 (sICAM-1) in active inflammatory bowel disease. Dig Dis Sci. 1996;39:1918–23. doi: 10.1007/BF02088125. [DOI] [PubMed] [Google Scholar]
- 30.Parkos CA, Colgan SP, Diamond MS, et al. Expression and polarization of intercellular adhesion molecule-1 on human intestinal epithelia: consequences for CD11b/CD18-mediated interaction with neutrophils. Mol Med. 1996;2:489–505. [PMC free article] [PubMed] [Google Scholar]
- 31.Panes J, Granger DN. Leucocyte–endothelial cell interactions: molecular mechanisms and implications in gastrointestinal disease. Gastroenterology. 1998;114:1066–90. doi: 10.1016/s0016-5085(98)70328-2. [DOI] [PubMed] [Google Scholar]
- 32.Yang H. Analysis of ICAM-1 gene polymorphism in immunologic subsets of inflammatory bowel disease. Exp Clin Immunogenet. 1997;14:214–25. [PubMed] [Google Scholar]
- 33.Shanahan F. The role of autoantibodies and autoimmunity in chronic inflammatory disorders of the gut. Curr Opin Gastroenterol. 1992;8:988–92. [Google Scholar]
- 34.Das KM, Squillante L, Robertson FM. Amplified expression of intercellular adhesion molecule-1 (ICAM-1) and M(r) 40K protein by DLD-1 colon tumor cells by interferon-gamma. Cell Immunol. 1993;147:215–21. doi: 10.1006/cimm.1993.1061. [DOI] [PubMed] [Google Scholar]
- 35.Strober W, Ehrhardt RO. Chronic intestinal inflammation: an unexpected outcome in cytokine or T cell receptor mutant mice. Cell. 1993;78:283–95. doi: 10.1016/0092-8674(93)80062-j. [DOI] [PubMed] [Google Scholar]
- 36.Cebra JJ, Jiang H, Šterzl J, Tlaskalova-Hogenová H. The role of mucosal microbiota in the development and maintenance of the mucosal immune system. In: Ogra P, Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, editors. Mucosal immunology. New York: Academic Press; 1999. pp. 267–80. [Google Scholar]
- 37.Andus T, Goebell H, Layer P, Scholmerich J. Dordrecht: Kluwer Ac. Publications; 1997. Inflammatory bowel diseases—from bench to bedside. [Google Scholar]
- 38.Powrie F. T cells in inflammatory bowel disease: protective and pathogenic roles. Immunity. 1995;3:171–4. doi: 10.1016/1074-7613(95)90086-1. [DOI] [PubMed] [Google Scholar]