

Abstract
We set out to determine whether inhibition of complement using sCR1 could influence the development and progression of collagen arthritis in the Lewis rat. Collagen arthritis was successfully established in the Lewis rat, using a novel immunization schedule. In separate experiments, cobra venom factor (CVF) and sCR1 were used to achieve systemic complement inhibition. Their respective effects on disease onset and on the progression of established disease compared with saline-treated control animals was explored. Arthritis was assessed by measurement of clinical score, paw diameter and paw volume. Complement inhibition using either CVF or sCR1, prior to the onset of clinical signs of inflammation, delayed the development of disease. CVF was ineffective in the treatment of established disease, whereas sCR1 delayed the progression of disease in affected joints and prevented the recruitment of further joints while the animals were complement-depleted. In the control saline-treated groups the disease continued to progress relentlessly. We conclude that complement activation is important in the initiation and maintenance of inflammation in collagen arthritis. The potent disease-modulating effect of sCR1 provides persuasive evidence that specific complement inhibiting agents may be an effective approach to the treatment of inflammatory joint diseases
Keywords: complement inhibition, animal models, sCR1
INTRODUCTION
Activation of complement is an important contributor to inflammation in rheumatoid arthritis (RA) [1]. Products of complement activation are present in abundance in the joint and plasma in RA and the levels of these products mirror disease activity [2–4]. The role of complement in the pathogenesis of animal models of human RA has clearly been demonstrated [5–8]. Immunofluorescence data in rats with collagen-induced arthritis (CIA) showed that the articular cartilage contained IgG and C3. When rats were depleted of complement with cobra venom factor (CVF), the onset of clinical disease was delayed and the development of disease in these animals correlated with the return of functional complement activity [6]. The use of CVF as a therapeutic agent in man, however, is not feasible, as CVF is highly immunogenic and promotes the generation of proinflammatory mediators (C3a, C5a and the membrane attack complex (MAC)) by activating the complement pathway to completion. The development of safe, specific and potent complement inhibitors such as sCR1 (TP10) has generated new interest in the potential for manipulation of the complement system in a range of inflammatory diseases [9–12]. sCR1, a potent inhibitor of the classical and alternative complement pathways, binds to C3b and C4b, resulting in inactivation of the C3 and C5 convertase. It has proved to be an excellent inhibitor of the in vitro and in vivo activation of serum complement in humans and a variety of other species. We have previously described its efficacy in antigen arthritis. Local inhibition of complement by intra-articular administration of sCR1 was effective in preventing disease in this model [8].
We describe here the effect of therapy with sCR1 on the clinical course and pathogenesis of CIA, a well-recognized rat model of human chronic synovitis. We show that intravenous (i.v.) sCR1 given either prior to the onset of disease or after the establishment of clinical disease had a potent anti-inflammatory effect. These results confirm that complement is involved in the generation of pathology in this disease model and that therapy with sCR1 offers a realistic prospect for treatment of human rheumatic disease.
MATERIALS AND METHODS
Animals
Inbred male Lewis rats (100–150 g) were obtained from Bantin and Kingman (The Field Station, Hull, UK) and housed in cages of four at the Biomedical Services Unit (UWCM, Cardiff, UK).
Collagen purification
Bovine nasal septa were obtained fresh from the local abattoir. Type II collagen was extracted from bovine nasal septa by pepsin digestion after previous treatment with 2 m magnesium chloride according to the method of Miller [13]. The extract was then extensively purified by differential salt precipitation and by dialysis against phosphate buffers. Collagen was stored lyophilized. Aliquots were dissolved overnight in 0.01 m acetic acid at 4°C before use as an immunogen.
Establishing the model
Initial experiments sought to determine the arthritogenicity of the collagen preparation and the nature and reproducibility of the arthritis produced. Using published protocols [5,14], results were unsatisfactory in that the incidence of disease achieved in our hands was low. We therefore developed a modified protocol as follows. Bovine type II collagen was dissolved in 0.01 m acetic acid at a concentration of 2 mg/ml by stirring at 4°C overnight. The collagen was emulsified at a ratio of 1:1 with Freund's incomplete adjuvant (FIA). Lewis rats were sensitized by inoculation with 0.5 ml of the emulsion (containing 500 μg of bovine type II collagen) intradermally at several sites on the back and the base of the tail (day 0). Seven days later all animals were re-immunized using an identical protocol.
Assessment of clinical disease
Rats were assessed daily for clinical signs of arthritis. A subjective clinical score was devised based on the degree of periarticular oedema and erythema as well as individual joint involvement and deformity. Each of the four limbs were graded as shown in Table 1. The severity of clinical symptoms was represented as the sum of the scores of all four limbs. The maximum score was therefore 20. A score of 8 or above represented severe disease.
Table 1.
Description of the macroscopic appearance of the individual arthritic paws of the animals and the clinical scores attributed to each paw

The severity of clinical symptoms was represented as the sum of the scores of all four limbs. The maximum score was therefore 20. A total score of ≥ 8 represented severe disease.
Additional indices of disease were obtained by measurement of paw diameters using a Mitutoyo digital calliper and paw volume by water displacement plesythmography. The water displacement plethysmography apparatus was constructed in house and was based on mercury displacement plethysmography. The apparatus had been calibrated in house and it had been shown that the weight of water displaced by an inflamed hind paw correlated with the clinical score attributed to that paw by a blinded observer. Results were expressed as the daily change in paw diameter (in mm) and paw volumes (in ml) from the measurement on the day of treatment. All measurements were done in triplicate by an observer unaware of the treatment regime and the mean of the three values was taken.
Functional activity of the complement inhibitory agents CVF and sCR1, determined by haemolytic assays
CVF
CVF was purified from the venom of the Thai cobra (Naja Naja kaouthia) (Sigma) by the method of Lachmann & Hobart [15] and was a gift from Dr J. P. Camilleri (UWCM).
Five Lewis rats were anaesthetized with halothane/oxygen. Blood (500 μl) was taken from the tail vein using a 26 G butterfly needle and plasma retained for assessment of baseline complement activity. All animals were then injected intraperitoneally with 100 U of CVF (day 0). All animals were bled daily from the tail vein under halothane anaesthesia for 7 consecutive days after the initial i.p. injection. The plasma was separated from the erythrocytes by centrifugation and aliquots of plasma were stored at −70°C until assayed.
sCR1
Recombinant, endotoxin-free sCR1 (TP10) at a concentration of 4.95 mg/ml in sterile PBS pH 7 was provided by Avant Immunotherapeutics Inc. (Needham, MA). At time 0, three naive animals were anaesthetized with halothane/oxygen. Blood (500 μl) was taken from the tail vein using a 26 G butterfly needle and plasma retained for assessment of baseline complement activity. All animals were then injected through the same needle with sCR1 (15 mg/kg). Blood samples (100 μl) were taken from the tail vein under anaesthesia at designated time points thereafter. The plasma was separated from the erythrocytes by centrifugation and aliquots of plasma were stored at −70°C until assayed. Twelve hours after the first dose of sCR1, a second identical dose was given via the tail vein and blood sampling continued at intervals.
Haemolytic assays
The haemolytic activity of each plasma sample was measured by minor modifications of published methods to assess complement inhibition [16]. Briefly, plasma samples were diluted in veronal buffer and incubated with a standard concentration of sheep erythrocytes sensitized with ambozeptor (rabbit anti-sheep erythrocyte globin; Behring, Hounslow, UK). Results were expressed as a percentage of normal rat plasma haemolytic activity in the same system.
Effect of complement inhibition on clinical disease
Prophylaxis
(i) Treatment with CVF. To examine the effects of decomplementation on the development of disease, rats were randomly divided into treatment and control groups (seven animals per group) 7 days after the first collagen immunization (day 7), at which time they received either 100 U of CVF or an equal volume of saline by i.p. injection.
(ii) Treatment with sCR1. Seven days after the first immunization, animals were randomly divided into two equal groups (five animals per group). One group received i.v. sCR1 (15 mg/kg per animal) twice daily via the tail vein at 12 h intervals for 5 days (days 7–11), whereas the control group received an equal volume of i.v. saline at the same time points.
Disease was assessed in each experiment by clinical score and by measurement of daily change in paw diameter and paw volume by an observer unaware of the treatment protocol.
Treatment of established disease
(i) CVF. Rats were immunized with bovine type II collagen as described above. Following the onset of clinical inflammation in one or more hind paws (approximately 12–14 days after the first immunization), animals were divided into two groups matched for clinical score. Each group consisted of five animals. One group received an i.p. injection of 100 U of CVF while the control group received an i.p. injection of saline.
(ii) sCR1. In a separate experiment, animals were again immunized with bovine type II collagen as described above. On day 14, all animals displaying clinical arthritis were divided into two groups, matched as closely as possible for clinical score (four animals per group). One group received sCR1 (15 mg/kg) by i.v. injection via the tail vein twice daily for 5 days (days 14–19), whilst the other received an equal volume of saline at the same dosing schedule.
Disease was again assessed by a blinded observer by clinical score, measurement of daily change in paw diameter and paw volume.
Statistical analysis
Student's t-test was used to assess the difference between the diameters, volumes and clinical scores of the treated and untreated animals. Non-parametric analyses were also performed where appropriate.
RESULTS
Establishing the model
The modified protocol described here enabled the establishment of a reproducible, and consistent model of inflammatory collagen arthritis in which 100% of animals developed disease. Twelve to 14 days after the primary immunization, animals began to show evidence of clinical inflammation in one or more hind paws. The first manifestation of disease was erythema of one or more ankle joints, followed by involvement of the metatarsal and interphalangeal joints. Table 2 shows the incidence of disease at different times in three different experiments.
Table 2.
The incidence of arthritis in three different experiments using the revised immunization protocol (two intradermal injections of bovine type II collagen a week apart on days 0 and 7)
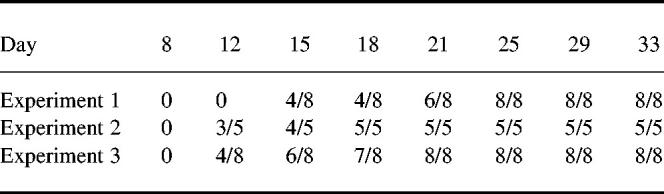
The typical time course of the development and progression of disease (as assessed by clinical score, paw diameter and paw volume) is shown in Fig. 1 (experiment 1, Table 2). By 24 days all animals showed evidence of disease, predominantly in the hind paws. The disease was always a progressive disease with joint recruitment following the same pattern: tarsal, metatarsophalangeal followed by the interphalangeal.
Fig. 1.
Typical time course of the development of collagen-induced arthritis (CIA) in male Lewis rats using the modified protocol. Disease was assessed by (a) clinical score, (b) paw diameter, and (c) paw volume. The results are mean ± s.e.m. (n = 8).
The interphalangeal joints were never solely involved, and inflammation in these joints was invariably associated with inflammation in the tarsal joint. Forepaws became inflamed at approximately 24 days after primary immunization; again the pattern of joint recruitment was carpal followed by the metacarpophalangeal joints and the interphalangeal joints. Not all animals developed forepaw inflammation and forepaw involvement never occurred in isolation without severe hind paw arthritis.
Complement inhibition delays onset of clinical disease in collagen arthritis
Rats were evaluated for the presence of arthritis at regular intervals following primary immunization. Disease severity was quantified by measurement of clinical score, paw diameter and paw volume.
CVF
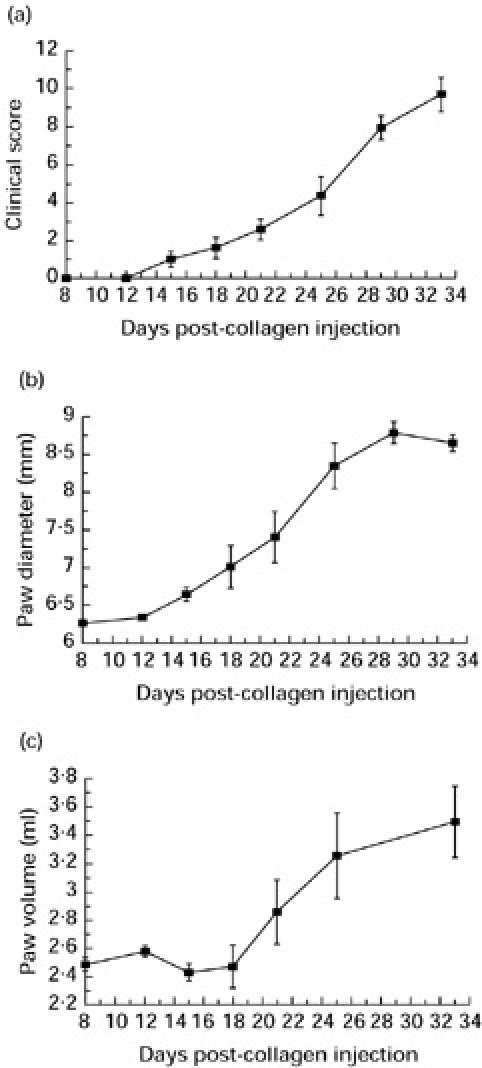
Treatment of rats with CVF on day 7 following the first collagen injection decreased the haemolytic activity of serum to zero. Complete decomplementation persisted for 5 days. The onset of clinical disease, manifest by swelling and erythema of multiple limbs, was delayed following complement depletion compared with saline-treated controls (Fig. 2a–c). Clinical disease was present in 6/7 of the control group at day 14 (mean clinical score 2.28 ± 0.93, mean change in paw diameter 1.94 ± 0.38 mm, mean change in paw volume 0.95 ± 0.184 ml). No clinical disease was apparent in the treated group at this time (mean clinical score 0, mean change in paw diameter 0.2 ± 0.04 mm, mean change in paw volume 0.13 ± 0.06 ml). Disease was first apparent on day 16 in the treated group. Disease was mild at this time, 5/7 animals had a score of 1 in one or more hind paw, whereas two animals had no evidence of inflammation in any paw. Significant differences were seen in all three parameters of disease activity (mean clinical score 2.0 ± 0.258 versus 5.33 ± 1.14 (P < 0.05)), change in mean paw diameter (1.05 ± 0.16 mm versus 2.368 ± 0.3 mm (P < 0.02)) and change in paw volume (0.464 ± 0.12 ml versus 1.04 ± 0.18 ml (P < 0.05)) at this time point. By day 18 post-collagen injection, however, all animals in treatment and control groups showed clinical disease of similar severity with no difference between any of the three parameters of disease activity.
Fig. 2.
The effect of cobra venom factor (CVF), given on day 7 prior to disease onset, on the development of collagen-induced arthritis (CIA) as measured by clinical score (a), change in paw diameter (b) and change in paw volume (c). Treated animals (n = 7) (▪) were decomplemented for 5 days (from day 7 to day 12), control animals (n = 7) (•) remained complement-sufficient. The period of decomplementation is represented by the solid bar. **Statistically significant difference between the control and treated groups, P < 0.02.
Haemolytic activity of the rat serum in the treated group had returned to normal by day 13. Decomplementation therefore delayed the clinical onset of disease by approximately the period for which the animals were complement-depleted, but when the animals became complement-replete the disease progressed as in the control group.
sCR1
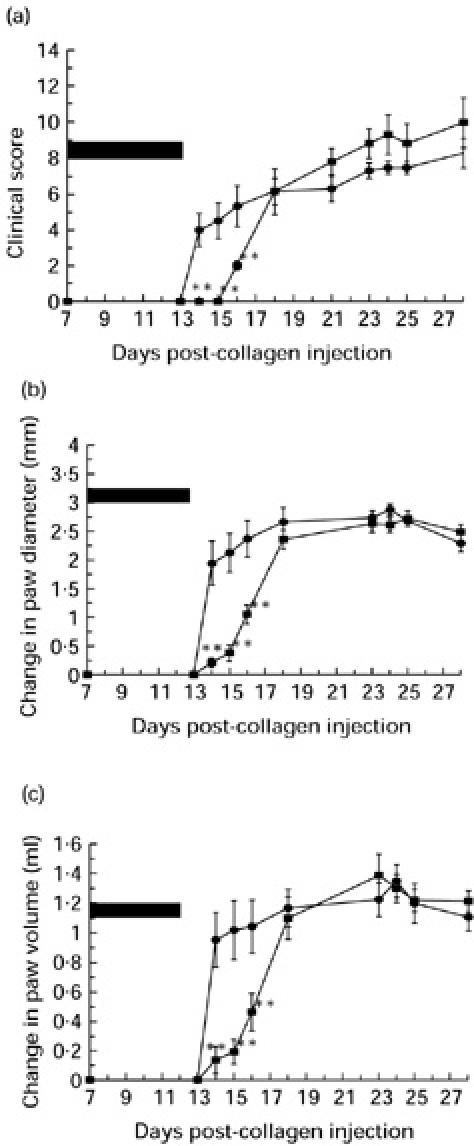
The effect of sCR1 therapy on the prevention of disease is shown in Fig. 3a–c. One group of animals received sCR1 (15 mg/kg per animal) twice daily from day 7 to day 12. Using this dosing schedule the haemolytic activity of rat plasma was maintained at a level < 20% of the activity of normal rat plasma during this 5-day period (data not shown). Similar to CVF therapy, treatment with sCR1 delayed the development of disease during the period of complement inhibition (Fig. 3a–c). Disease was first apparent in the control group on day 13 post-collagen injection with 3/5 animals showing some degree of paw swelling. By day 15, all of the control animals showed clinical signs of disease (mean total clinical score 2.4). Disease first emerged at day 16 in the treated group, with mild disease in one animal only (a score of 1 in one hind paw). Joint recruitment and inflammation progressed gradually thereafter. The incidence and severity of the arthritis in the paws of the animals in the treated and untreated groups remained significantly different during the first 20 days. Comparing the mean clinical scores between the treated and untreated control animals a significant difference was seen on days 15–20 (day 15, 0 versus 2.4 ± 0.67, P < 0.01; day 16, 2 ± 0.2 versus 4.4 ± 0.51, P < 0.002; day 17, 1 ± 0.61 versus 5.4 ± 0.4, P < 0.002; day 19, 2.2 ± 1.29 versus 6.2 ± 0.5, P < 0.05; day 20, 3.2 ± 1.39 versus 7.2 ± 0.5, P < 0.05). Thereafter the differences between the groups became non-significant, although the trend of lower scores in the treated animals persisted.
Fig. 3.
The effect of intravenous sCR1 on the development of collagen-induced arthritis (CIA) as assessed by clinical score (a), change in paw diameter (b) and change in paw volume (c). Treated animals (▪) (n = 5) were given i.v. sCR1 every 12 h for 5 consecutive days starting on day 7 in order to maintain complete complement inhibition. Control animals (•) (n = 5) received a twice daily injection of i.v. saline. The period of complement inhibition is represented by the solid bar. ***Statistically significant difference between the control and treated groups, P < 0.002; **statistically significant difference between the control and treated groups, P < 0.02; *statistically significant difference between the control and treated groups, P < 0.05.
Similar results were seen in the differences in the change in paw diameters and change in paw volumes (Fig. 3b,c). Significant differences were seen for the change in paw diameters from days 13–20 (day 13, −0.111 ± 0.157 mm versus 0.175 ± 0.157 mm, P < 0.02; day 15, −0.152 ± 0.121 mm versus 1.107 ± 0.299 mm, P < 0.002; day 18, 0.026 ± 0.13 mm versus 1.704 ± 0.0306 mm, P < 0.001; day 19, 1.81 ± 0.364 mm versus 2.755 ± 0.228 mm, P < 0.002; day 20, 2.21 ± 0.44 mm versus 3.187 ± 0.11 mm, P < 0.02) and similarly in the paw volumes between days 16 and 20 (day 16, 0.097 ± 0.08 ml versus 0.362 ± 0.0.107 ml, P < 0.05; day 18, −0.006 ± 0.06 ml versus 0.796 ± 0.107 ml, P < 0.002; day 20, 0.635 ± 0.15 ml versus 1.236 ± 0.06 ml, P < 0.002). The differences thereafter were non-significant, although again the trend of lower indices persisted.
Complement inhibition reduces arthritis in established disease
CVF
Animals that showed evidence of arthritis in one or more hind paws on day 14 were matched for clinical score and divided into two groups (five animals per group). One group was treated with i.p. CVF and the other with i.p. saline. The results were disappointing, as only on day 18 was the difference in mean clinical score between the groups significant (1.6 ± 0.92 versus 3.75 ± 1.03, P < 0.05). The trend of lower scores persisted in the treated animals until day 22, but the difference did not reach statistical significance after day 18. CVF had a potent anti-inflammatory effect in one animal, the clinical score decreasing from 2 to 0; this was maintained between days 15 and 22. By day 26, all animals showed evidence of disease, and all hind paws were noticeably swollen. The differences between the paw diameters and paw volumes of the control and treated groups never reached statistical significance.
sCR1
On day 14, animals manifesting signs of disease in one or more hind paws were divided into two groups matched as closely as possible for clinical score (four animals per group). From days 14–19, animals in one group received twice daily an i.v. dose of sCR1 (15 mg/kg), whereas those in the other group received an equal volume of i.v. saline. The differences in the clinical scores, paw diameters and paw volumes are shown in Fig. 4a–c.
Fig. 4.
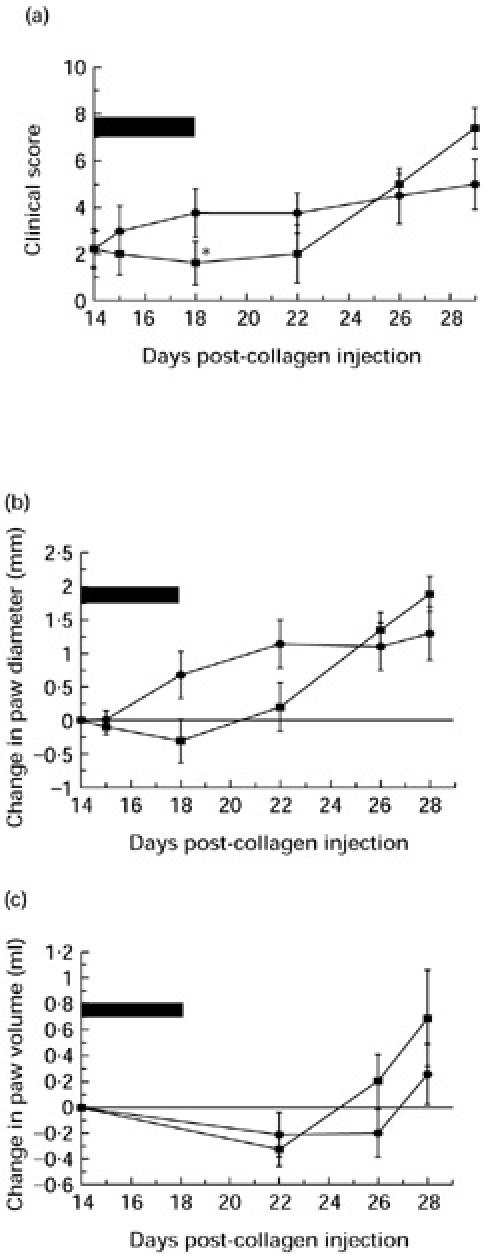
The effect of cobra venom factor (CVF) on established collagen-induced arthritis (CIA) as assessed by clinical score (a), change in paw diameter (b) and change in paw volume (c). On day 14, arthritic animals were divided into two equal groups matched for clinical score. Treated animals (▪) received a single i.p. dose of CVF, the animals were decomplemented between days 14 and 18 (as represented by the solid black bar). Control animals (•) received an i.p. dose of saline on day 14 and remained complement-sufficient throughout. *Statistically significant difference between the treated and untreated groups, P < 0.05.
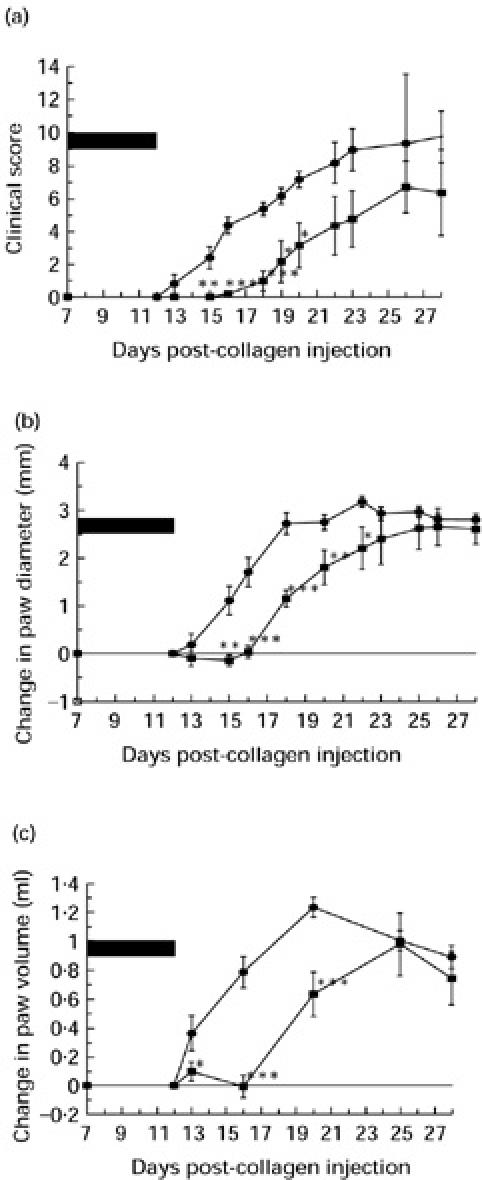
Intravenous administration of sCR1 had a marked inhibitory effect on the progression of disease as assessed by clinical score (Fig. 4a). The difference reached statistical significance (P < 0.05) between days 16 and 20 but the trend was maintained throughout the course of the experiment.
Figure 4b,c show the effect on the change in paw diameters and paw volumes. Disease activity was expressed as the change from the value on the day of treatment. The trend in paw diameter measurements was identical to that observed for clinical score; therapy with sCR1 improved the paw diameters, the change in diameter reaching statistical significance (P < 0.05) at days 16–18. The differences in paw volumes, despite showing a similar trend, however, failed to reach statistical significance. On days 15–18 all three parameters of disease had improved from the previous days with a maximum effect at day 16. At day 16 the maximum differences between the treated and control groups were seen (clinical score 1.5 ± 0.87 versus 4.67 ± 0.67; diameter −0.42 ± 0.02 mm versus 0.68 ± 0.26 mm; volume 1.78 ± 0.09 ml versus 2.24 ± 0.13 ml). This reflected the attenuation of swelling in joints already involved at initiation of treatment as well as slowing of disease spread to additional joints.
DISCUSSION
RA is a chronic disease of unknown aetiology that affects predominantly synovial joints. The current treatment of RA is non-specific. The presently available so-called ‘disease-modifying agents' are unsatisfactory in that they are poorly effective and have not been shown to reduce significantly the destructive component of disease. Major advances in the understanding of the pathogenic mechanisms of RA and in the development of therapies have been made through the use of animal models. Experimental CIA is a well recognized model of chronic peripheral polyarthritis which was serendipitously discovered by Trentham et al. in 1977 during attempts to raise a reference antiserum in rats to collagen [17]. The histopathology of CIA is characterized by a proliferative synovitis and pannus formation that erodes the adjacent cartilage and ultimately produces severe articular injury and ankylosis [18,19]. CIA shares clinical, histological and immunological features with human RA [20–22] and has therefore been regarded as an important model of this human disease. CIA probably represents an example of immune complex-mediated tissue injury. It has been suggested that rats immunized with type II collagen mount a humoral response and synthesize circulating antibodies to type II collagen. Some of these antibodies cross-react with native rat articular cartilage collagen and bind, leading to the formation of localized intra-articular immune complexes with consequent activation of complement [23]. Complement activation products can cause damage to structures within the joint either directly via the MAC or indirectly through C5a-mediated recruitment and activation of phagocytic cells. CIA has already proved useful in identifying novel therapeutic agents which decrease joint damage [24–26], and the role of complement in the pathogenesis of CIA has already been demonstrated [6,26], making this an appropriate model in which to explore the therapeutic efficacy of novel anti-complement agents such as sCR1.
The initial aim of the experiments detailed here was to establish the model in a reliable, reproducible manner. Attempts to induce CIA by closely adhering to two published protocols [23,27] failed, provoking us to develop a modified protocol which induced disease in 100% of animals and proved very reproducible. The clinical features of the arthritis produced using the modified protocol (two intradermal injections a week apart) resembled the arthritis previously reported by others [6,14,17]. However, the disease produced was less explosive than that reported previously with joints recruited sequentially. The order of recruitment always followed the same pattern in hind paws: tarsal followed by metatarsal followed by interphalangeal joints. The incidence of forepaw involvement was approximately 30–50% in each experiment. Forepaw involvement was invariably seen in the animals that displayed the most severe hind paw arthritis. Forepaws were never solely involved. The sequential rather than explosive nature of disease was advantageous, as this provided a larger window in which to show an anti-inflammatory effect of therapy. The consistently high incidence, with 100% of the animals in each experiment developing disease (Table 2), contrasted with that reported using published protocols in inbred (28% incidence) and outbred (40%) Wistar [22,28] and outbred Sprague Dawley (75%) strain rats [23]. The basis for these differences is not clear, although it is likely that they are at least in part genetically determined [29]. The increased incidence was again advantageous, in that fewer animals were required per group to test therapeutic efficacy.
Previously published studies have utilized CVF to show that CIA was complement-dependent [5,6]. For this reason, in all our experiments CVF was used as a comparator for the effects of sCR1. As previously reported, decomplementation using CVF prior to onset of disease delayed the onset of clinical signs for 2 or 3 days, the length of time required for recovery of complement levels in serum (Fig. 2). Treatment with sCR1, starting prior to the onset of disease and continuing for 5 days, also delayed disease onset for at least 3 days, and disease, as judged by clinical score, was significantly less than in the control group for a further 3 days (Fig. 3). Therapy with sCR1 could not be continued longer because of the development of a strong immune response, as previously described [10,11]. The results show that sCR1 had a more potent disease-inhibiting effect than CVF. This could be explained by the fact that CVF causes decomplementation by activating the pathway to completion with the consequent release of proinflammatory mediators, whereas complement inhibition by sCR1 is non-depleting, acting by inhibiting the C3 and C5 convertases [9,27,30,31].
Of greater relevance to the treatment of human RA is the part played by complement in propagation of established disease. It is a criticism of many experimental therapies in animal models that treatment is initiated prior to the onset of clinical disease and effects on the established inflammatory response are not known. Only in one previous study has an effect of complement inhibition on established joint disease been reported. Wang and colleagues showed that systemic administration of a blocking anti-C5 MoAb effectively prevented the development of arthritis in the murine model of collagen arthritis and was also effective in ameliorating established disease [26].
In the experiments described here, animals were treated on day 14 in the course of CIA, at which time disease was already established. Decomplementation with CVF prevented the progression of established disease in affected joints and also prevented the recruitment of further joints while the animals were complement depleted, whereas in the control groups the disease continued to progress relentlessly (Fig. 4). A potent anti-inflammatory effect was seen in some paws with scores of 1 and 2, but decomplementation did not reverse established disease in paws with scores of ≥ 3. When established CIA was treated with sCR1 from day 14 for 5 days, a more potent disease-modulating effect was observed (Fig. 5). The clinical score, the most consistent measure of disease severity, was significantly reduced compared with control animals throughout the treatment period and remained low through to the end of the experiment on day 28 (Fig. 5a). Measurement of the paw diameter (Fig. 5b) and volume (Fig. 5c) did not provide such clear evidence of efficacy, because the most severely ankylosed joints, which achieve high clinical score, were often less swollen than early arthritic joints which have a greater degree of soft tissue oedema.
Fig. 5.
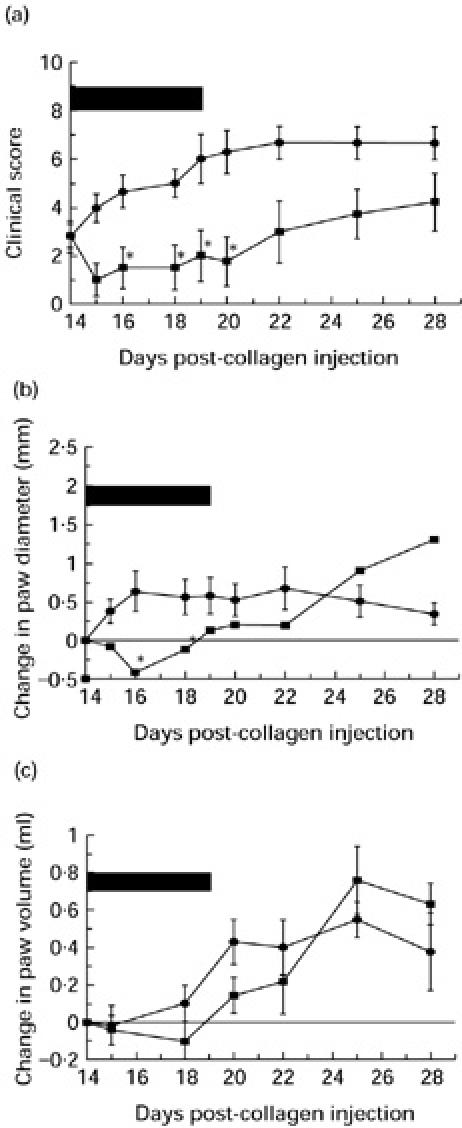
The effect of sCR1 on established collagen-induced arthritis (CIA) as assessed by clinical score (a), change in paw diameter (b) and change in paw volume (c). On day 14, arthritic animals were divided into two equal groups matched for clinical score. Treated animals (▪) received intravenous sCR1 every 12 h for 5 consecutive days from day 14 to day 18. The period of complement inhibition is represented by the solid black bar. Control animals (•) received a twice daily intravenous dose of saline for 5 consecutive days from day 14 and remained complement-sufficient throughout. *Statistically significant difference between the treated and untreated groups, P < 0.05.
The results reported here confirm that complement is important in both the initiation and propagation of disease in the CIA model of inflammatory arthritis in the rat. Prior therapy with sCR1 for 4 days delayed disease onset and reduced severity of disease, and therapy of established arthritis stopped further progression of disease and reversed pathologic changes in joints exhibiting mild arthritis. Therapy with sCR1 could be given for a maximum of only 5 days because of the immunogenicity of this human protein in rats. Therapy with sCR1 in humans will not be subject to the same time constraints and sCR1 is known to be a far better inhibitor of human than rat complement [9–12]. It is thus likely that sCR1 will be a much more effective agent in man. Taken together, these findings, along with our earlier studies in antigen arthritis [8] and the work of others in mouse models of arthritis [32], provide strong support for testing sCR1 and other specific complement-inhibiting agents in therapy of inflammatory joint disease in man.
Acknowledgments
R.M.G. would like to thank the ARC for financial support during this study (grant no, 0507).
REFERENCES
- 1.Morgan BP. Complement, clinical aspects and relevance to disease. London: Academic Press; 1990. The biological effects of complement activation; pp. 130–40. [Google Scholar]
- 2.Morgan BP, Daniels RH, Williams BD. Measurement of terminal complement complexes in rheumatoid arthritis. Clin Exp Immunol. 1988;73:473–8. [PMC free article] [PubMed] [Google Scholar]
- 3.Sanders ME, Kopicky JA, Wigley FM, Shin ML, Frank MM, Joiner KA. Membrane attack complex of complement in rheumatoid synovial tissue demonstrated by immunofluorescent microscopy. J Rheumatol. 1986;13:1028–34. [PubMed] [Google Scholar]
- 4.Kemp PA, Spragg JH, Brown JC, Morgan BP, Gunn CA, Taylor PW. Immunohistochemical determination of complement activation in joint tissues of patients with rheumatoid arthritis and osteoarthritis using neo-antigen specific monoclonal antibodies. J Clin Lab Immunol. 1992;37:147–62. [PubMed] [Google Scholar]
- 5.Morgan K, Clague RB, Shaw MJ, Firth SA, Twose TM, Lennox Holt PJ. Native type 2 collagen induced arthritis in the rat. The effect of complement depletion by cobra venom factor. Arthritis Rheum. 1981;24:1356–62. doi: 10.1002/art.1780241104. [DOI] [PubMed] [Google Scholar]
- 6.Kerwar SS, Bauman N, Cronsky AL, Sloboda AE. Studies on type II collagen induced polyarthritis in rats effect of complement depletion. J Immunopharmacol. 1982;3:323–37. doi: 10.3109/08923978109031065. [DOI] [PubMed] [Google Scholar]
- 7.Schwab JH, Allen JB, Anderle SK, Dallorf F, Eisenberg R, Cromartie WJ. Relationship of complement to experimental arthritis induced in rats by streptococcal cell walls. Immunology. 1982;46:83–7. [PMC free article] [PubMed] [Google Scholar]
- 8.Goodfellow RM, Williams AS, Levin JL, Williams BD, Morgan BP. Local therapy with soluble complement receptor one (sCR1) suppresses inflammation in rat mono-articular arthritis. Clin Exp Immunol. 1997;110:45–52. doi: 10.1046/j.1365-2249.1997.5111408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weisman MF, Barlow T, Leppo MK, et al. Soluble human complement receptor type 1. In vivo inhibitor of complement suppressing post ischaemic myocardial inflammation and necrosis. Science. 1990;249:146–51. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 10.Piddlesden SJ, Storch MK, Hobbs M, Freeman AM, Lassmann H, Morgan BP. Soluble recombinant complement receptor 1 inhibits inflammation and demyelination in antibody mediated demyelinating experimental allergic encephalomyelitis (ADEAE) J Immunol. 1994;152:5477–84. [PubMed] [Google Scholar]
- 11.Pratt JR, Hibbs MJ, Laver AJ, Smith RAG, Sacks SH. Effects of complement inhibition with soluble complement receptor-1 on vascular injury and inflammation during renal allograft rejection in the rat. Am J Pathol. 1996;149:2055–66. [PMC free article] [PubMed] [Google Scholar]
- 12.Couser WG, Johnson RJ, Young BA, Yeh CG, Toth CA, Rudolph AR. The effects of soluble recombinant complement receptor 1 on complement mediated experimental glomerulonephritis. J Am Soc Nephrol. 1995;5:1888–94. doi: 10.1681/ASN.V5111888. [DOI] [PubMed] [Google Scholar]
- 13.Miller EJ. Structural studies on cartilage collagen employing limited cleavage and solubilisation by pepsin. Biochemistry. 1972;11:4903–9. doi: 10.1021/bi00776a005. [DOI] [PubMed] [Google Scholar]
- 14.Stuart JM, Cremer MA, Townes AS, Kang AH. Type II collagen-induced arthritis in rats: passive transfer with serum and evidence that IgG anticollagen antibodies can cause arthritis. J Exp Med. 1982;155:1–16. doi: 10.1084/jem.155.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lachmann PJ, Hobart MJ. Complement technology. In: Weir DM, editor. Handbook of experimental immunology, Chapter 5A. 3. Vol. 1. Oxford: Blackwell Scientific Publications; 1978. [Google Scholar]
- 16.Whaley K. Measurement of complement. In: Whaley K, editor. Methods in complement for clinical immunologists. Edinburgh: Churchill-Livingstone; 1985. pp. 77–139. [Google Scholar]
- 17.Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen: an experimental model of arthritis. J Exp Med. 1977;146:857–67. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caulfield JP, Hein A, Dynesius-Trentham R, Trentham DE. Morphologic demonstration of two stages in the development of type II collagen-induced arthritis. Lab Invest. 1982;46:321–43. [PubMed] [Google Scholar]
- 19.Terato K, Hashida R, Miyamoto K, et al. Histological, immunological and biochemical studies on type II collagen-induced arthritis in rats. Biochem Res. 1982;3:495–505. [Google Scholar]
- 20.Steffen C, Timpl R. Antigenicity of collagen and its application in the serological investigation of rheumatoid arthritis sera. Int Arch Allergy Appl Immunol. 1963;22:333–40. doi: 10.1159/000229376. [DOI] [PubMed] [Google Scholar]
- 21.Andriopoulos NA, Mestecky J, Miller GJ, Bradley EL. Antibodies to native and denatured collagens in sera of patients with rheumatoid arthritis. Arthritis Rheum. 1976;19:613–7. doi: 10.1002/art.1780190314. [DOI] [PubMed] [Google Scholar]
- 22.Trentham DE, Townes AS, Kang AH, David JR. Humoral and cellular sensitivity to collagen in type II collagen-induced arthritis in rats. J Clin Invest. 1978;61:89–96. doi: 10.1172/JCI108929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan K, Clague RB, Shaw MJ, Holt PJL. Native type II collagen-induced arthritis in the rat I. Incidence and humoral response to collagen. Ann Rheum Dis. 1980;39:285–90. doi: 10.1136/ard.39.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sloboda AE, Birnbaum JE, Oronsky AL, Kerwar SS. Studies on type II collagen induced polyarthritis in rats: effect of antiinflammatory and antirheumatic drugs. Arthritis Rheum. 1981;24:616–23. doi: 10.1002/art.1780240408. [DOI] [PubMed] [Google Scholar]
- 25.Stuart JM, Myers LK, Townes AS, Kang AH. Effect of cyclophosphamide, hydrocortisone and levamisole on collagen-induced arthritis in rats. Arthritis Rheum. 1981;24:790–4. doi: 10.1002/art.1780240606. [DOI] [PubMed] [Google Scholar]
- 26.Breban M, Fournier C, Gougerot-Pocidalo M, Muffat-Joly M, Pocidalo J. Protective effects of ciprofloxacin against type II collagen induced arthritis in rats. J Rheumatol. 1992;19:216–22. [PubMed] [Google Scholar]
- 27.Staines NA, Hardingham T, Smith M, Henderson B. Collagen-induced arthritis in the rat: modification of immune responses by free collagen and immune anti-collagen antiserum. Immunology. 1981;44:737–44. [PMC free article] [PubMed] [Google Scholar]
- 28.Clague RB, Morgan K, Shaw MH, Holt PLJ. Native type II collagen induced arthritis in rats. II Relationship between the humoral immune response to native type II collagen and arthritis. J Rheumatol. 1980;7:775–81. [PubMed] [Google Scholar]
- 29.Griffiths MM, Eichwald EJ, Martin JH, Smith CB, DeWitt CW. Immunogenetic control of experimental type II collagen-induced arthritis I. Susceptibility and resistance among inbred strains of rats. Arthritis Rheum. 1981;24:781–9. doi: 10.1002/art.1780240605. [DOI] [PubMed] [Google Scholar]
- 30.Furthmayr H, Timpl R. International review of connective tissue research. Vol. 7. New York: Academic Press; 1976. Immunochemistry of collagens and procollagens; pp. 61–99. [DOI] [PubMed] [Google Scholar]
- 31.Knight SC, Mertin J, Stackpole A, Clarke JC. Induction of immune responses in vivo with small numbers of veiled (dendritic) cells. Proc Natl Acad Sci USA. 1983;80:6032–5. doi: 10.1073/pnas.80.19.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc Natl Acad Sci USA. 1995;92:8955–9. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]