

Abstract
Recent studies demonstrate in vivo and in vitro cytokine dysregulation in CF epithelial cells. To see if these abnormalities may be generalized to other cells expressing cystic fibrosis transmembrane conductance regulator (CFTR) but not directly exposed to local inflammation, we studied mRNA transcription, intracellular protein production and extracellular secretion of IL-2, IL-4, IL-5, IL-10 and interferon-gamma (IFN-γ) from freshly isolated blood mononuclear and CD4+ T cells from CF patients and controls. Cells were activated by phorbol myristate acetate (PMA) and anti-CD3, PMA–ionomycin, or lipopolysaccharide (LPS) and assessed for cytokine mRNA transcription by semiquantitative reverse transcriptase-polymerase chain reaction, intracellular protein production by flow cytometry, and secretion by supernatant ELISA. Cytokine expression was highly stimulus-dependent. CF cells showed higher IL-10 transcription than control cells after maximal activation by LPS (P = 0·01); despite this, cytokine production and secretion were equivalent to controls. CF cells showed lower cellular IL-10 production after PMA-anti-CD3 activation (P = 0·002). CF cells secreted less IFN-γ than control cells after maximal activation by PMA-anti-CD3 (1836 ± 273 pg/ml versus 9635 ± 3437 pg/ml, P = 0·04). IL-2, IL-4 and IL-5 regulation was similar to controls. We conclude that CF mononuclear cells show selective cytokine dysregulation after maximal activation, namely reduced IFN-γ secretion and increased IL-10 mRNA without increased production or secretion. These findings extend defects described in respiratory epithelial cells to circulating immunoregulatory cells, suggesting a link between CF genotype and cytokine dysregulation.
Keywords: cystic fibrosis, inflammation, cytokines, interferon, interleukin
Introduction
Recent studies of presymptomatic infants with CF, diagnosed by newborn screening, have suggested the early post-natal onset of airways inflammation, including in some cases before any evidence of microbial infection. Airways inflammation in CF is characterized even in these infants by elevated numbers and proportions of neutrophils and their products, high levels of the proinflammatory cytokines IL-8, IL-1β, and tumour necrosis factor-alpha (TNF-α) [1,2]. Other investigators have reported that airway cells and fluids from CF mutant mice as well as CF patients exhibit exaggerated proinflammatory responses to activating stimuli, which may reflect an intrinsic dysregulation or disruption of cytokine homeostasis, including low levels of the anti-inflammatory cytokine IL-10 [3–6]. This has been called an endogenous pathway to inflammation that may precede and/or amplify inflammation due to the exogenous pathway of microbial infection [7].
Lymphocytes are known to functionally express cystic fibrosis transmembrane conductance regulator (CFTR); in patients with CF, CFTR-regulated chloride channel function is impaired similarly to epithelial cells [8–10]. We reported that T cell clones derived from peripheral blood lymphocytes (PBL) of CF patients exhibit an altered cytokine secretion profile with an impaired IL-10 secretory response after activation by lectin binding or cross-linkage of the T cell receptor with anti-CD3 in the presence of phorbol myristate acetate (PMA) [11]. These results suggested that an intrinsic cytokine dysregulation may exist in immunoregulatory cells functionally expressing mutant CFTR.
Because T cell clone phenotype and behaviour are markedly influenced by the methodology and conditions of cell culture, we have studied cytokine responses from fresh peripheral blood mononuclear cells (PBMC) obtained from patients with CF and controls in order to examine further whether there is an underlying dysregulation of cytokine secretion independent of microenvironmental inflammatory effects present in the airways. We examined the in vitro effect of activating stimuli on peripheral immunoinflammatory cells from CF patients and healthy controls with regard to mRNA transcription, intracellular cytokine production, and extracellular cytokine secretion. These stimuli act upon mononuclear and/or T cells via distinct signal transduction pathways: PMA in combination with the Ca2+ ionophore ionomycin via Ca2+ influx, PMA in combination with anti-CD3 via T cell receptor ligation and cross-linkage, and lipopolysaccharide (LPS) via ligation of the glycosylphosphatidylinositol-anchored membrane protein CD14. Our results indicate a variety of differences in regulation of cytokine expression after activation at the transcriptional, translational, and secretory levels between CF and control immunoregulatory cells.
SUBJECTS and METHODS
Subjects
Peripheral blood (40–50 ml) was obtained by venepuncture from adult non-smoking subjects. The study subjects included 16 healthy adult laboratory personnel (six males, 10 females, ages 24–47 years) and 24 patients with CF (13 males, 11 females, age 27 ± 11 years (mean ± s.d.), range 11–53 years). All patients with CF had diagnosis confirmed by pilocarpine iontophoresis sweat test and were pancreatic insufficient with pancreatic enzyme treatment to control steatorrhoea. The CF patients were in stable clinical condition, were not suffering from pulmonary exacerbation, and were not receiving systemic corticosteroids, with the exception of one 25-year-old female on long-term prednisone 10 mg po qod for treatment of allergic bronchopulmonary aspergillosis whose blood was obtained on the ‘off’ prednisone day. All CF patients were on chronic suppressive antibiotic therapy for chronic bacterial endobronchitis with inhaled tobramycin 300 mg bid, colistin 150 mg bid, or oral ciprofloxacin 750 mg bid at the time of study. Pulmonary status varied widely, with forced expiratory volume (FEV)1% predicted at time of study ranging from 19% to 101% (48 ± 25%). All but one patient (who was infected with Alcaligenes xylosoxidans) were chronically infected with Pseudomonas aeruginosa. All patients were genotyped for 31 CFTR mutations. Eleven patients (46%) were homozygous for the δ F508 mutation. Except for one patient who was heterozygous for G542x, the remainder (50%) were heterozygous for δ F508, with other identified mutations including 3659delC (n = 1), G85E (n = 2), W1282X (n = 1), 1898 + 1 (n = 1), 2184delA (n = 1), and G542X (n = 1); six patients carried an unidentified mutation at the second allele. The study was approved by the Stanford University Human Subjects Committee and informed consent was obtained from participants.
Cell culture and cytokine induction
PBMC were isolated by Histopaque-1077 (Sigma, St Louis, MO) density gradient centrifugation, washed with PBS, and resuspended in AIM-V serum-free lymphocyte culture medium (Life Technologies/Gibco; catalogue no. 12055-091)−1% heat-inactivated human AB serum. For experiments involving activation with PMA–anti-CD3, magnetizable polystyrene beads coated with monoclonal anti-human CD4 (Dynabeads; Dynal, Oslo, Norway) were used to isolate CD4+ T cells from PBMC. Isolated T cells or PBMC were dispensed into 24-well culture plates in 1-ml aliquots of 1 × 106 cells/well. Cells were cultured in duplicate at 37°C in a humidified atmosphere containing 5% CO2 in the presence of one of the following activators: (i) 1 ng/ml PMA and 500 ng/ml ionomycin; (ii) PMA−10 μg/ml immobilized anti-human CD3; or (iii) 0·1 mg/ml LPS. Monoclonal anti-CD3 was immobilized by coating microtitre plate wells with 10 μg/ml anti-CD3 for 1 h at 37°C and washing twice with PBS. At 6, 24 or 48 h after incubation, cells and supernatants were harvested. Brefeldin 10 μg/ml was added 2–4 h before harvesting for flow cytometric intracellular protein analysis (see below) [12].
RNA isolation
Total RNA was isolated from pelleted cell preparations with TRIzol reagent (Gibco BRL). After phase separation with chloroform and centrifugation, the RNA was precipitated with isopropyl alcohol and washed with 75% ethanol. The final RNA pellet was dissolved in Rnase-free water and concentration determined by UV spectrophotometry.
First-strand cDNA was generated from 1 mg total RNA using AMV-reverse transcriptase and oligo(dT) priming according to the manufacturer's instructions (cDNA Cycle kit; Invitrogen). As part of cDNA synthesis, a ‘minus-reverse transcriptase’ (m-RT) negative control was prepared from each RNA sample. Generation of a polymerase chain reaction (PCR) product from this control indicates contamination by genomic DNA. Positive control cDNA was generated from the MS2 test RNA and random primers supplied in the kit.
Specific primers for β-actin, interferon-gamma (IFN-γ), IL-4 and IL-10 were used as specified by the manufacturer (Clontech). The expected amplified PCR fragments were: IFN-γ, 427 bp; IL-4, 344; IL-10, 328; and β-actin, 838. The internal MS2 primers frame a 900 base pair fragment of the MS2 RNA.
A PCR master mix was prepared with final concentration (per PCR reaction) of 10 mm Tris–HCl, pH 8·3; 50 mm KCl; 1·5 mm MgCl2; 0·2 mm each of dATP, dCTP, dGTP, and dTTP (dNTPs); and 2 U of Taq DNA polymerase (Gibco BRL). The master mix was pipetted into thin-walled tubes to which 0·4 mm of each cytokine-specific oligonucleotide primer was added. The RT reaction was diluted 1:5 and 2 μl added to each tube. The cDNA generated from MS2 test RNA was amplified using internal MS2 primers (Invitrogen). The negative control was 2 μl water in place of a RT product. The m-RT controls were amplified using β-actin primers. Only β-actin primers amplified genomic DNA to produce a PCR product the same size as the specified cDNA product. Reactions were done in 50-μl volumes. The temperature cycling programme was 95°C for 2 min for two cycles; 95°C for 45 s, 55°C for 45 s, and 72°C for 60 s for 35 cycles, with a final extension at 72°C for 10 min in a thermal cycler (Hybrid Omnigene).
RT-PCR products were diluted using 6x loading dye mix and electrophoresed in a 70-V constant-voltage field in 2% agarose containing ethidium bromide. Electrophoresis continued until the bromophenol blue dye front had migrated 6 cm. The gel was visualized using a UV light box and Polaroid photographs were taken as a permanent record. Agarose gels were scanned using a BioRad Gel Doc 1000 on a MacIntosh Power PC with the Molecular Analyst 2.1.1 program (BioRad)[13]. Identical gating was employed for each scan and the mean of three readings was used for each value recorded.
Cytoplasmic cytokine production assayed by flow cytometry
After harvesting and activation as described above, cells were fixed in 2% formaldehyde and then permeabilized in 1% bovine serum albumin (BSA)–PBS−0·5% saponin [14]. Cells were incubated with the following dye-conjugated cytokine-specific MoAbs (PharMingen): mouse anti-human IFN-γ–FITC clone 4SB3 (0·25 µ g/106 cells), mouse anti-human IL-4–PE clone 8D4-8 (0·5 μg/106 cells), or rat anti-human IL-10–PE clone JES-9D7 (5 μg/106 cells). For IL-2 detection, a two-step process was used: rat anti-human IL-2 (clone 17H12, kindly provided by Dr H. Yssel of DNAX, Palo Alto CA), followed by goat anti-rat FITC diluted 1:150 in 1% BSA (Caltag). Isotype-matched irrelevant antibody or unlabelled autologous antibody blocking (30 min preincubation at room temperature before addition of labelled antibody) were used as controls. IL-10 isotype control antibody was a rat anti-human IgG1–PE used at 5 μg/106 cells (PharMingen). For IFN-γ isotype control, mouse anti-human IFN-γ (2·5 μg/106 cells) was preincubated with fixed-permeabilized cells before adding mouse anti-human IFN-γ–FITC antibody. Flow cytometry was performed in a FACScan flow cytometer (Becton Dickinson) with filter settings at 530 nm (for FITC) or 585 nm (for PE) to read the stained cells. Cells (1 × 105) were collected in open gate and analysed with FACScan software Lysis II Version 1.0.
Cytokine secretion by sandwich ELISA
Monoclonal antibody ELISAs were performed as previously described [11]. Briefly, microtitre plate wells were coated with primary cytokine-specific capture antibodies overnight at 4°C. Cell culture supernatant samples and cytokine standards (provided by H. Yssel) were diluted in RPMI 1640 in 10% fetal calf serum (FCS) and incubated overnight at 4°C. Detecting antibodies were derivatized with the haptenic group nitroiodophenylacetate (NIP) [15]. The secondary NIP-conjugated antibodies were then incubated for 2 h at room temperature. Wells were then incubated with immunoperoxidase-conjugated anti-NIP MoAb for 1 h at room temperature. Enzyme substrate o-phenylenediamine (0·5 mg/ml) dissolved in citric-phosphate buffer with fresh H2O2 was added to develop the plate. The reaction was stopped with 5 n sulphuric acid and results determined colorimetrically by optical density (OD) reading at 495 nm using an automated ELISA reader. The sensitivity of the ELISA was approx. 50 pg/ml for each cytokine. The cytokine ELISA had an intra-assay coefficient of variation (CV) of < 5–10% (n = 5) and an interassay CV of 8–21% (n = 3–4).
Statistical analysis
Differences between mean values were compared by Student's unpaired two-tailed t-tests. Variances are expressed as s.e.m. unless otherwise indicated. Significance was set at a two-tailed P value of 0·05.
Results
Cytokine transcription after activation
Transcription of cytokine mRNA after activation was assessed in isolated CD4+ T cells or PBMC by RT-PCR. As shown in Fig. 1, kinetics of cytokine transcription after activation varied according to activation stimulus. The calcium ionophore ionomycin in combination with phorbol ester resulted in strong early stimulation of T cell IFN-γ, IL-2, and IL-4, but only a weak IL-10 response (Fig. 1a). In contrast, activation by phorbol ester and cross-link ligation of the T cell receptor via immobilized anti-CD3 MoAb resulted in strong early, transient, and relatively selective activation of IL-10 (Fig. 1b). A third pattern of activation was observed using LPS as the activating stimulus for PBMC. With LPS there was a strong selective early activation of IL-10 (Fig. 1c); this persisted for at least 48 h (Fig. 1d).
Fig. 1.
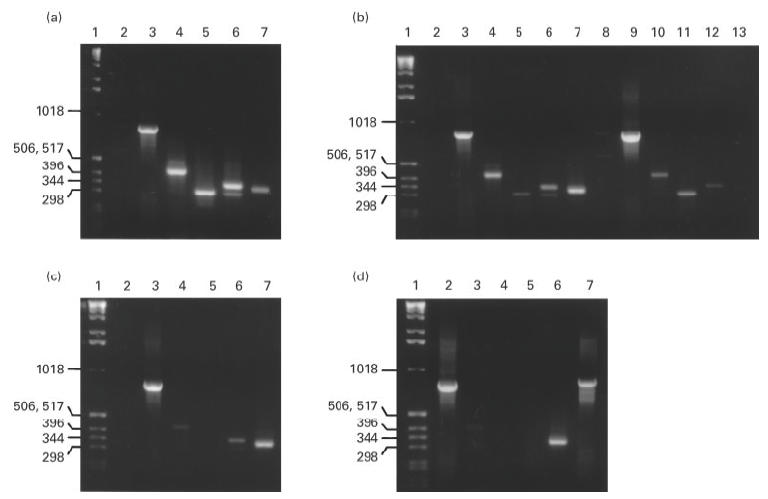
Reverse transcriptase-polymerase chain reaction (RT-PCR) kinetics of cytokine mRNA transcription by T cells after activation is shown after activation with several stimuli. (a) Activation by phorbol myristate acetate (PMA)–ionomycin for 6 h. Lane 4, IFN-γ; lane 5, IL-2; lane 6, IL-4; lane 7, IL-10. (b) Activation by PMA–anti-CD3. Lanes 1–7 are with a 6-h incubation and lanes 8–13 are with a 24-h incubation. Here, IFN-γ is shown in lanes 4 and 10, IL-2 in lanes 5 and 11, IL-4 in lanes 6 and 12, and IL-10 in lanes 7 and 13. (c,d) Activation by lipopolysaccharide (LPS) for 6 h and 48 h, respectively. The IL-10 bands are in (c) lane 7 and (d) lane 6. Standard weight markers are shown in lane 1 for all panels. Negative H2O control is in lane 2 in (a–c). Constitutively produced β-actin control is in lane 3 in (a–c) and lane 2 in (d). Positive test RNA control is shown in (d) lane 7.
Transcription of cytokine genes in CF and normal cells was compared. CF and normal CD4+ T cells responded similarly to activation with phorbol ester and anti-CD3 with respect to cytokine gene transcription. Figure 2 shows a representative comparison of RT-PCR results. T cell IFN-γ and IL-10 responses following PMA–anti-CD3 were compared in seven CF and 17 control subjects. CF subjects showed a trend toward lower IFN-γ responses compared with controls (mean ± s.d. 49 ± 32% versus 92 ± 53% of β-actin levels, respectively; P = 0·06). IL-10 mRNA transcription after PMA–anti-CD3 were comparable in CF and control T cells (mean ± s.d. 45 ± 25% versus 63 ± 47% in CF and controls, respectively). IL-4 and IL-5 mRNA expression was also similar in the two groups (data not shown).
Fig. 2.
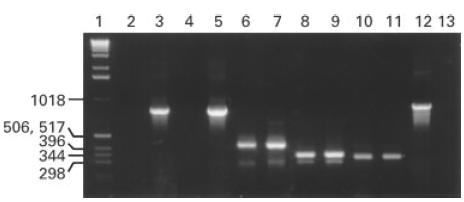
Transcription of cytokine genes by T cells from a CF patient and a normal control was compared after 6 h activation with phorbol myristate acetate (PMA)–anti-CD3. Lane 1, kD weight standard markers; lanes 2 and 4, negative controls from the CF patient and the control subject, respectively; lanes 3 and 5, β-actin; lanes 6 and 7, CF and control IFN-γ; lanes 8 and 9, CF and control IL-4; lanes 10 and 11, CF and control IL-10. Lane 12 shows positive control (test RNA) and lane 13 shows negative (H2O) control.
After activation by LPS, β-actin gene expression was identical in PBMC from 15 CF and 15 control subjects (27 681 ± 2974 densitometry units (DU) versus 27 875 ± 2002 DU, respectively). However, PBMC from CF patients showed a higher level of IL-10 mRNA transcription than controls after activation by LPS (IL-10 percentage of β-actin gene expression = 92 ± 82% (mean ± s.d.) in CF versus 29 ± 30% in controls, P = 0·01) (Fig. 3).
Fig. 3.
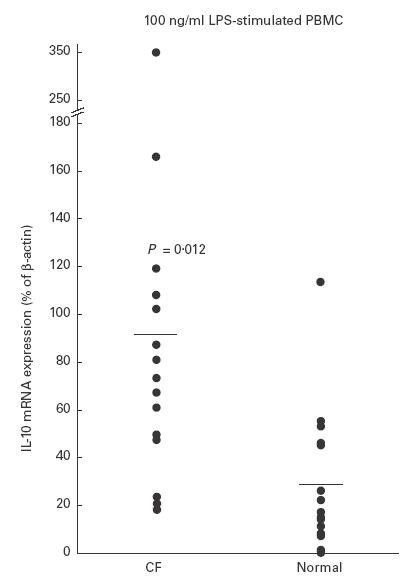
IL-10 mRNA transcription expressed as percent of constitutive β-actin mRNA transcription in CF and control peripheral blood mononuclear cells after activation with lipopolysaccharide (LPS).
Intracellular cytokine production
Intracellular cytokine production was assessed immunohistochemically by flow cytometry of activated cells which were then fixed with formaldehyde, permeabilized with saponin, and stained with cytokine-specific fluorochrome-conjugated MoAbs.
Unstimulated PBMC were double-labelled with antibodies specific either for T cells (CD3–FITC) or monocytes (CD33–FITC) and anti-IL-10–PE to assess whether monocytes contributed to IL-10 production in the lymphocyte-gated cell population studied in flow cytometry. These studies confirmed that comparable low percentages (0·3–1·0% of control and 0·7–2·1% of CF) of IL-10-producing CD33+ monocytes were present in lymphocyte-gated test samples. A 2–20% range (2·9–8·5% of control and 1·8–19·2% of CF) of CD3+ T cells produced IL-10.
After stimulation of T cells by PMA–anti-CD3, similar proportions of CF (n = 10) and control (n = 10) derived T cells were positive for cytoplasmic IFN-γ (1·9 ± 0·4% versus 4·0 ± 1·3%, respectively). Fewer T cells from CF patients were IL-10+ than controls (9·0 ± 1·8% versus 21·6 ± 3·0%, respectively; P = 0·002). In contrast, after maximal stimulation by LPS, comparable percentages of CF (n = 26) and control (n = 16) PBMC were positive both for cytoplasmic IFN-γ (2·6 ± 1·9% of CF and 0·8 ± 0·4% of control cells positive) and IL-10 (15·1 ± 3·5% of CF and 15·3 ± 3·5% of control cells positive).
T cell responses to activation with phorbol ester and the Ca2+ ionophore ionomycin were compared with those following activation with phorbol ester and ligation of the T cell receptor through anti-CD3. Ca2+ influx was found to be a powerful and comparable inducer of IFN-γ and IL-2 production in both control and CF-derived T cells (Fig. 4). Similar results were found with IL-4 (data not shown). These results indicate that Ca+2 entry is a strong inducer of IFN-γ, IL-2, and IL-4 production by T cells, and that CF cells respond similarly to control cells for these cytokines. A distinct pattern was seen for IL-10 responses. In control T cells, maximal Ca2+ entry after cell activation with ionomycin inhibited IL-10 production. CF T cells were less responsive to activation with anti-CD3 and also less sensitive to the inhibitory effect of ionomycin (Fig. 5).
Fig. 4.
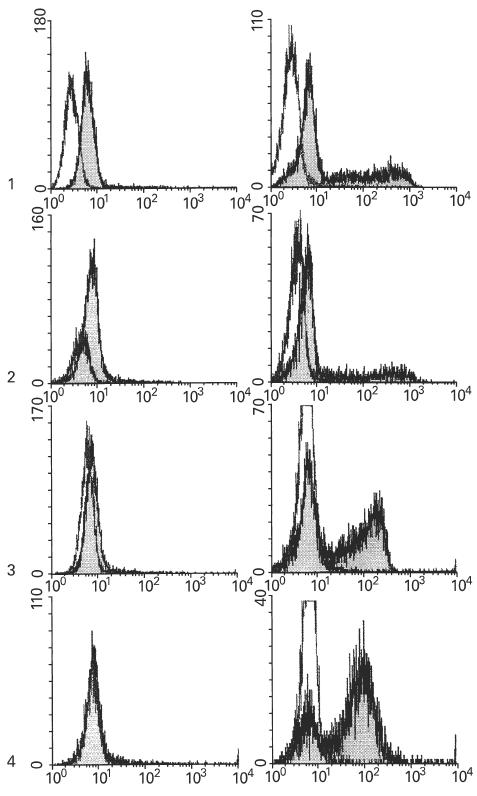
Flow cytometry showing cytoplasmic cytokine production by T cells after activation by phorbol myristate acetate (PMA)–ionomycin in comparison with activation by PMA–anti-CD3. Top graphs (1): IFN-γ production by control cells with anti-CD3 or ionomycin after 6 h activation. Mean fluorescence intensity ratio (Fir) of stimulated cells (shaded histogram) to unstimulated cells (clear histogram) was 2·2 after activation with anti-CD3 (left) and 28·2 after activation with ionomycin (right). The second pair of graphs (2) shows respective IFN-γ results with CF cells. The FIr after anti-CD3 (left) or ionomycin (right) was 1·6 and 20·1, respectively. The third pair of graphs (3) shows results for IL-2 with control cells. The FIr after anti-CD3 (left) or ionomycin (right) was 0·2 and 11·1, respectively. The final pair of graphs (4) shows results for IL-2 with CF cells. The FIr after anti-CD3 (left) or ionomycin (right) was 0 and 12·7, respectively.
Fig. 5.
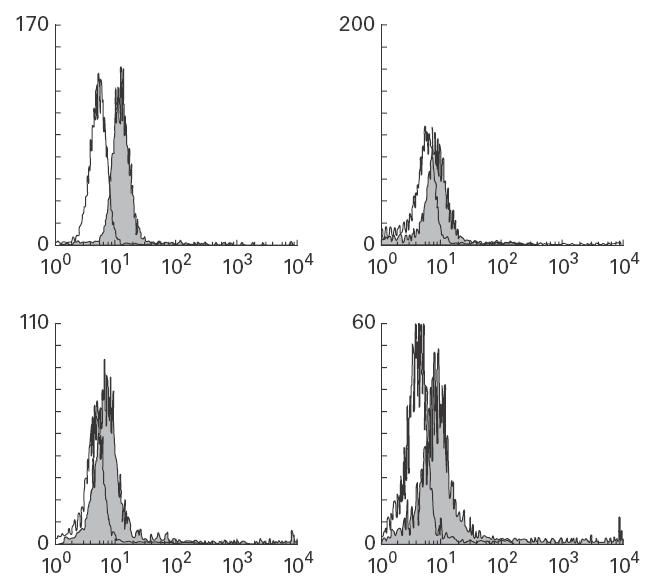
Flow cytometry showing IL-10 production by T cells after stimulation with phorbol myristate acetate (PMA)–anti-CD3 or ionomycin. Top, IL-10 production by control T cells after activation with PMA–anti-CD3 (left) or ionomycin (right). The mean fluorescence intensity ratios (Fir) were 2·4 and 1·5 for anti-CD3 and ionomycin, respectively. Bottom, results with CF T cells after stimulation with anti-CD3 (left) or ionomycin (right). The FIr was 1·5 and 2·2 for anti-CD3 and ionomycin, respectively.
Cytokine secretion
Extracellular cytokine secretion was measured in cell culture supernatants after activation of T cells with phorbol ester and anti-CD3, or PBMC with LPS (Table 1). After PMA–anti-CD3 activation, large amounts of IFN-γ were secreted by control T cells, averaging nearly 10 000 pg/ml. T cells from CF patients (n = 10) secreted significantly lower levels of IFN-γ than cells from controls (n = 10) after activation with phorbol ester–anti-CD3 (1836 ± 273 pg/ml versus 9635 ± 3437 pg/ml, respectively; P = 0·04). Much lower but readily detectable amounts of IL-10 were secreted by T cells, averaging about 800 pg/ml. CF and control T cells secreted comparable levels of IL-10 (1154 ± 358 versus 693 ± 187, respectively). There were also no significant differences between CF and control T cells in levels of IL-5 (217 ± 46 pg/ml versus 543 ± 160 pg/ml, respectively). IL-4 levels were below detection limits (< 50 pg/ml) in both the CF and control groups.
Table 1.
Cytokine secretion of CD4+ T cells after activation by phorbol ester–anti-CD3 and peripheral blood mononuclear cells after activation by lipopolysaccharide (LPS)
| Cytokine | Activator | CF | (n) | Control | (n) |
|---|---|---|---|---|---|
| IL-4 | PMA/α CD3 | 11 ± 3 | (10) | 31 ± 9 | (10) |
| IL-5 | PMA/α CD3 | 217 ± 46 | (10) | 543 ± 160 | (10) |
| IL-10 | PMA/α CD3 | 1154 ± 358 | (10) | 693 ± 187 | (10) |
| LPS | 2701 ± 479 | (26) | 1951 ± 434 | (16) | |
| IFN-γ | PMA/α CD3 | 1836 ± 273 | (10) | 9635 ± 3437* | (10) |
| LPS | 103 ± 27 | (26) | 129 ± 24 | (16) |
Results in pg/ml, mean ± s.e.m.
P = 0·04, CF versus control.
LPS was the most potent as well as selective inducer of IL-10 secretion in PBMC. After activation with LPS, PBMC from CF patients (n = 26) secreted comparable amounts of IL-10 to cells from controls (n = 16) (2701 ± 479 pg/ml versus 1951 ± 434 pg/ml, respectively). Activation of PBMC with LPS resulted in minimal but comparable IFN-γ secretion, averaging approx. 100 pg/ml, in CF and control PBMC (103 ± 27 pg/ml versus 129 ± 24 pg/ml, respectively). As after PMA–anti-CD3 activation, we did not see stimulation of IL-4 secretion by LPS in either group.
Discussion
Inflammation is recognized as a major pathogenic component of CF lung disease. Current understanding of this process acknowledges the early onset, severity, and persistence of inflammatory changes in airway tissue and fluids of CF patients [1–3]. However, the origin of inflammation is controversial. Conventionally, inflammatory changes have been considered a consequence of early onset of respiratory epithelial infection with microbial pathogens. A number of recent studies examining respiratory epithelial fluids and cultured epithelial cells support this view [16–18]. However, a growing body of evidence has emerged supporting the possibility that inflammatory changes in CF may precede infection and may be triggered by non-infectious stimuli or even represent a basal feature of cells involved in inflammation, including epithelial cells, submucosal gland serous cells, mononuclear cells, and granulocytes [1,2,4–6,19–29]. The present study was conducted in order to test the hypothesis that activation of inflammatory cells by a variety of stimuli would uncover differences in cytokine regulation between cells from patients with CF and controls.
For these studies, we chose to use freshly isolated PBMC and CD4+ T cells. Previously, we had found that cloned T cells from CF patients exhibited a deficiency in IL-10 secretion after activation, but could not conclude from these data whether this represents an inherent property of the T cells or an influence of methods of cell cloning [11]. Our rationale for studying freshly isolated PBMC and T cells was based on a desire to use cells that, on the one hand, are not subject to the intensely inflamed environment of the CF airway (where secondary changes confound interpretation), and, on the other hand, are not subject to alterations that may emerge from long-term cell culture and cloning. It is well established that these cells functionally express CFTR [8–11]. In addition, practical considerations in studying enough subjects to yield adequate sample sizes militate against collection of cells obtained from bronchoalveolar lavage. Moreover, preliminary studies using lavaged airway cells have revealed technical problems related to presence of infection and mucus contamination that interfere with assay techniques, in particular flow cytometry (unpublished observations).
In comparing CF and control PBMC and T cells, it was necessary to establish the pattern of cytokine response to various activating stimuli. For these experiments we utilized three activators: (i) PMA–anti-CD3 activation of T cells, acting through ligation of the T cell receptor; (ii) PMA–ionomycin activation of T cells, acting through extracellular Ca2+ influx; and (iii) LPS activation of PBMC, acting through the LPS receptor signal transduction pathway. Activation was assessed at the three levels of mRNA transcription by RT-PCR; cytoplasmic protein production by flow cytometry of fixed, permeabilized and stained cells using cytokine-specific MoAbs; and ELISA of supernatants from short-term cultures of stimulated cells. These studies showed the following: (i) ionomycin activation results in strong expression of IFN-γ, IL-2 and IL-4 genes; (ii) anti-CD3 activation results in strong, early and relatively specific expression of IL-10; (iii) LPS activation results in strong, specific and persistent expression of IL-10 (Fig. 1). Thus, cytokine expression is strongly dependent on a specific activation stimulus, and results must be interpreted accordingly. Our data support those previously reported in this regard [30–34].
When CF cells were compared with controls, the most striking finding was a markedly reduced IFN-γ secretory response following activation with anti-CD3, a stimulus normally inducing a maximal IFN-γ response. CF cells produced a mean of approx. 2 ng/ml IFN-γ after anti-CD3 activation, compared with approx. 10 ng/ml in control cells. No significant differences in IL-10 secretion, or IL-2 (data not shown), IL-4 or IL-5 secretion were found. The reduction in IFN-γ secretion did not appear to be solely attributable to reduced gene transcription or intracellular protein production, although CF cells did show a trend towards reduced IFN-γ mRNA transcription following PMA–anti-CD3.
This finding, a marked reduction in IFN-γ secretion upon activation by CF cells compared with controls, could have important pathogenic implications. In an animal model of chronic Ps. aeruginosa endobronchitis, it was shown that IFN-γ can reduce the inflammatory response [35]. Moreover, vaccination with a conjugated Ps. aeruginosa alginate that shifts the immune response towards a more prominent IFN-γ response was associated with reduced disease and mortality [36]. Finally, strains of mice producing higher IFN-γ responses had reduced disease and mortality [37]. If CF is also characterized by a deficiency in IFN-γ response, this could contribute to the pathogenesis of chronic pulmonary infection with Ps. aeruginosa and suggests a new therapeutic approach to increase endobronchial IFN-γ levels, either directly, e.g. with aerosolized IFN-γ, or indirectly, e.g. with IL-12 immunization [38,39].
A recently described feature of CF lung disease is deficiency of epithelial cell-associated and bronchial lavage fluid IL-10 [6,40,41]. Since IL-10 is a critical homeostatic anti-inflammatory cytokine, the cause of this deficiency and its role in pathogenesis are important issues in deciphering the pathogenesis of CF. Our previous results employing cloned peripheral blood T cells suggested an intrinsic deficiency in IL-10 secretion in CF [11]. The studies presented here with freshly isolated PBMC and T cells suggest that CF cells can mount normal IL-10 responses when maximally stimulated. The observed deficiency of IL-10 production in CF bronchoalveolar lavage fluid (BALF) and brushed respiratory epithelial cells does not appear to be due to a generalized inability of CF cells to express this cytokine, but rather may be related to microenvironmental effects such as down-regulation by other cytokines.
We were surprised to find that CF PBMC showed significantly greater IL-10 mRNA transcription that control cells after LPS activation (Fig. 3). However, no greater cytoplasmic production or extracellular secretion of IL-10 by CF cells was noted. In fact, a lower proportion of CF cells produced cell-associated immunoreactive IL-10 than controls after anti-CD3 activation as assessed by flow cytometry. It is possible that this paradox may be due to a difference between CF and control cells in response to Ca2+ activation. T cell IL-10 secretion is down-regulated by increased Ca2+ influx [30,33]. It has been proposed that lymphocyte Cl− channels participate in regulation of Ca2+ influx and thus of cell activation and signal transduction [42]. If CF cells are impaired in this regulatory mechanism they may be subject to altered extracellular Ca2+ entry, which could in turn affect IL-10 expression [30,33]. Results with flow cytometry suggest this may be the case, as CF cells showed less Ca2+-mediated inhibition of IL-10 production compared with control cells after activation with ionomycin (Fig. 5). We are currently exploring the hypothesis that chloride flux alterations can regulate cellular cytokine expression [43].
Our results raise a number of questions that require further study. Our patients, the youngest of whom was aged 11 years, had long-established CF lung disease including chronic bacterial endobronchitis. It is not known if our findings can be extended to younger patients or those not chronically infected with pathogenic Gram-negative bacteria. We did not investigate the possible effect of circulating cytokines or receptors on the blood cells we employed for our studies, which were freshly obtained, washed, and stimulated in short-term cell cultures. Despite these uncertainties, our findings are consistent with the general argument that CF cells display inherent aberrations in cytokine regulation that may be a consequence of altered CFTR function. Reductions in IFN-γ without compensatory increases in IL-10 may prime CF tissues to exaggerated, prolonged, and severe inflammation. These findings suggest that cytokine responses may represent valuable biologic surrogates of correction of CFTR dysfunction, e.g. by gene transfer [44,45], and also suggest that new therapeutic approaches augmenting pulmonary IFN-γ and IL-10 responses may modify the inflammation that determines the course of cystic fibrosis lung disease.
Acknowledgments
Portions of this work were presented at meetings of the American Thoracic Society (Am J Resp Crit Care Med 1997; 155:A198) and the American Academy of Allergy, Asthma and Immunology (J Allergy Clin Immunol 1997; 99:S330). This work was supported by research grants from the Cystic Fibrosis Foundation, CFRI, and the Ross Mosier Fund.
REFERENCES
- 1.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso F, Riches Dwh. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–82. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- 2.Balough KR, McCubbin M, Weinberger M, Smits W, Ahrens R, Fick R. The relationship between infection and inflammation in the early stages of lung disease in cystic fibrosis. Pediatr Pulmonol. 1995;20:63–70. doi: 10.1002/ppul.1950200203. [DOI] [PubMed] [Google Scholar]
- 3.Heerecken AV, Walenga R, Konstan MW, Bonfield T, Davis PB, Ferkol T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest. 1997;100:2840–5. doi: 10.1172/JCI119828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noah TL, Blach HR, Cheng PW, Wood RE, Leigh Mw. Nasal and bronchoalveolar lavage cytokines in early cystic fibrosis. J Infect Dis. 1997;175:638–47. doi: 10.1093/infdis/175.3.638. [DOI] [PubMed] [Google Scholar]
- 5.Tabary O, Zahm JM, Hinnrasky J, et al. Selective up-regulation of chemokine IL-8 expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am J Physiol. 1998;153:921–30. doi: 10.1016/S0002-9440(10)65633-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonfield TL, Konstan MW, Berger M. Altered respiratory epithelial cell cytokine production in cystic fibrosis. J Allergy Clin Immunol. 1999;104:72–78. doi: 10.1016/s0091-6749(99)70116-8. [DOI] [PubMed] [Google Scholar]
- 7.Moss Rb. Pathways to inflammation in cystic fibrosis. Pediatr Pulmonol. 1996;13(Suppl.):158–60. [Google Scholar]
- 8.Chen JH, Schulman H, Gardner P. A cAMP-regulated chloride channel in lymphocytes that is affected in cystic fibrosis. Science. 1989;243:657–60. doi: 10.1126/science.2464852. [DOI] [PubMed] [Google Scholar]
- 9.McDonald TV, Nghiem PT, Gardner P, Martens Cl. Human lymphocytes transcribe the cystic fibrosis transmembrane conductance regulator gene and exhibit CF-defective cAMP-regulated chloride current. J Biol Chem. 1992;267:3242–8. [PubMed] [Google Scholar]
- 10.Dong Y, Chao AC, Kouyama K, Hsu YP, Bocian RC, Moss RB, Gardner P. Activation of CFTR chloride channel by nitric oxide in human T lymphocytes. EMBO J. 1995;14:2700–7. doi: 10.1002/j.1460-2075.1995.tb07270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moss RB, Bocian RC, Hsu YP, Dong Y, Wei T, Kemna M, Gardner P. Reduced IL-10 secretion by CD4+ T lymphocytes expressing mutant cystic fibrosis transmembrane conductance regulator. Clin Exp Immunol. 1996;106:374–88. doi: 10.1046/j.1365-2249.1996.d01-826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Misumi Y, Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem. 1986;261:11398–403. [PubMed] [Google Scholar]
- 13.Shea Tb. An inexpensive densitometric analysis system using a MacIntosh computer and a desktop scanner. Biotechniques. 1994;16:1126–8. [PubMed] [Google Scholar]
- 14.Jung T, Schauer U, Heusser C, Neumann C, Rieger C. Detection of intracellular cytokines by flow cytometry. J Immunol Methods. 1993;159:197–207. doi: 10.1016/0022-1759(93)90158-4. [DOI] [PubMed] [Google Scholar]
- 15.Immunoenzymatic assay of mouse and human cytokines using NIP-labeled anti-cytokine antibodies. In: Abrams Js., editor; Coligan J, editor. Current protocols in immunology. Suppl 13. New York: Wiley-Interscience; 1995. p. 6.20. [DOI] [PubMed] [Google Scholar]
- 16.Bedard M, McClure CD, Schiller NL, Francoeur C, Cantin A, Denis M. Release of interleukin-8, interleukin-6, and colony-stimulating factors by upper airway epithelial cells: implications for cystic fibrosis. Am J Respir Cell Mol Biol. 1993;9:455–62. doi: 10.1165/ajrcmb/9.4.455. [DOI] [PubMed] [Google Scholar]
- 17.Armstrong DS, Grimwood K, Carlin JB, et al. Lower airways inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med. 1997;156:1197–204. doi: 10.1164/ajrccm.156.4.96-11058. [DOI] [PubMed] [Google Scholar]
- 18.Black HR, Yankaskas JR, Johnson LG, Noah Tl. Interleukin-8 production by cystic fibrosis nasal epithelial cells after tumor necrosis factor-α and respiratory syncytial virus stimulation. Am J Respir Cell Mol Biol. 1998;19:210–5. doi: 10.1165/ajrcmb.19.2.3053. [DOI] [PubMed] [Google Scholar]
- 19.Jones MM, Seillheimer DK, Pier GB, Rosen Rd. Increased elastase secretion by peripheral blood monocytes in cystic fibrosis patients. Clin Exp Immunol. 1990;80:344–9. doi: 10.1111/j.1365-2249.1990.tb03290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Regelmann WE, Skubitz KM, Herron Jm. Increased monocyte oxidase activity in cystic fibrosis homozygotes and heterozygotes. Am J Respir Cell Mol Biol. 1991;5:27–33. doi: 10.1165/ajrcmb/5.1.27. [DOI] [PubMed] [Google Scholar]
- 21.Dagli E, Wamer JA, Besley CR, Warner Jo. Raised serum soluble interleukin-2 receptor concentrations in cystic fibrosis patients with and without evidence of lung disease. Arch Dis Child. 1992;67:479–81. doi: 10.1136/adc.67.4.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfeiffer KD, Hueksteadt TP, Hoidal Jr. Expression and regulation of tumor necrosis factor in macrophages from cystic fibrosis patients. Am J Respir Cell Mol Biol. 1993;9:511–9. doi: 10.1165/ajrcmb/9.5.511. [DOI] [PubMed] [Google Scholar]
- 23.Koller DY, Urbanek R, Gotz M. Increased degranulation of eosinophil and neutrophil granulocytes in cystic fibrosis. Am J Respir Crit Care Med. 1995;152:629–33. doi: 10.1164/ajrccm.152.2.7633718. [DOI] [PubMed] [Google Scholar]
- 24.DiMango EM, Zar HJ, Bryan R, Prince A. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J Clin Invest. 1995;96:2204–10. doi: 10.1172/JCI118275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witko-Sarsat V, Allen RC, Paulais M, Nguyen AT, Bessou G, Lenoir G, Descamps-Latcha B. Disturbed myeloperoxidase-dependent activity of neutrophils in cystic fibrosis homozygotes and heterozygotes, and its correction by amiloride. J Immunol. 1996;157:2728–35. [PubMed] [Google Scholar]
- 26.Kammouni W, Figarella C, Marchand S, Merten M. Altered cytokine production by cystic fibrosis tracheal gland serous cells. Infect Immun. 1997;65:5176–83. doi: 10.1128/iai.65.12.5176-5183.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zahm JM, Gaillard D, Dupuit F, Hinnrasky J, Porteous D, Dorin JR, Puchelle E. Early alterations in airway mucociliary clearance and inflammation of the lamina propria in CF mice. Am J Physiol. 1997;272:C853–9. doi: 10.1152/ajpcell.1997.272.3.C853. [DOI] [PubMed] [Google Scholar]
- 28.DiMango E, Ratner AJ, Bryan R, Tabibi S, Prince A. Activation of NF-κB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis epithelial cells. J Clin Invest. 1998;101:2598–605. doi: 10.1172/JCI2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell KJ, McRedmond J, Mukherji N, et al. Neutrophil adhesion molecule surface expression and responsiveness in cystic fibrosis. Am J Respir Crit Care Med. 1998;157:756–61. doi: 10.1164/ajrccm.157.3.9704008. [DOI] [PubMed] [Google Scholar]
- 30.Yssel H, De Waal Malefyt R, Roncarolo MG, Abrams JS, Lahesmaa R, Spits H, de Vries Je. IL-10 is produced by subsets of human CD4+ T cell clones and peripheral blood T cells. J Immunol. 1992;149:2378–84. [PubMed] [Google Scholar]
- 31.Gonzalez S, Beck L, Wilson N, Spiegelberg Hl. Comparison of interferon-γ and interleukin-4 production by peripheral blood mononuclear cells and isolated T cells after activation with polyclonal T cell activators. J Clin Lab Anal. 1994;8:277–83. doi: 10.1002/jcla.1860080506. [DOI] [PubMed] [Google Scholar]
- 32.Borish L, Aarons A, Rumbyrt J, Cvietusa P, Negri J, Wenzel S. Interleukin-10 regulation in normal subjects, patients with asthma. J Allergy Clin Immunol. 1996;97:1288–96. doi: 10.1016/s0091-6749(96)70197-5. [DOI] [PubMed] [Google Scholar]
- 33.Harada Y, Watanabe S, Yssel H, Arai K. Factors affecting the cytokine production of human T cells stimulated by different modes of activation. J Allergy Clin Immunol. 1996;98:S161–73. doi: 10.1016/s0091-6749(96)70063-5. [DOI] [PubMed] [Google Scholar]
- 34.Lai CKW, Ho S, Chan CHS, Chan J, Choy D, Leung R, Lai K. Cytokine gene expression profile of circulating CD4+ T cells in active pulmonary tuberculosis. Chest. 1997;111:606–11. doi: 10.1378/chest.111.3.606. [DOI] [PubMed] [Google Scholar]
- 35.Johansen HK, Hougen HP, Rygaard J, Hoiby N. Interferon-gamma treatment decreases the inflammatory response in chronic Pseudomonas aeruginosa pneumonia in rats. Clin Exp Immunol. 1996;103:212–8. doi: 10.1046/j.1365-2249.1996.d01-618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johansen HK, Cryz Sj, Jr, Hougen HP, Moser C, Hoiby N. Vaccination promotes TH1-like inflammation and survival in chronic Pseudomonas aeruginosa pneumonia. A new prophylactic principle. Behring Inst Mitt. 1997;98:269–73. [PubMed] [Google Scholar]
- 37.Moser C, Johansen HJ, Song Z, Hougen HP, Rygaard J, Hoiby N. Chronic Pseudomonas aeruginosa lung infection is more severe in Th2 responding BALB/c mice compared to Th 1 responding C3H/HeN mice. APMIS. 1997;105:838–42. [PubMed] [Google Scholar]
- 38.Boguniewicz M, Martin RJ, Martin D, Gibson U, Celniker A, Williams M, Leung Dy. The effects of nebulized recombinant interferon-gamma in asthmatic airways. J Allergy Clin Immunol. 1995;95:133–5. doi: 10.1016/s0091-6749(95)70162-1. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Trinchieri G, Wilson Jm. Recombinant IL-12 prevents formation of blocking IgA antibodies to recombinant adenovirus and allows repeated gene therapy to mouse lung. Nature Med. 1995;1:890–3. doi: 10.1038/nm0995-890. [DOI] [PubMed] [Google Scholar]
- 40.Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, Berger M. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152:2111–8. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- 41.Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hilliard JB, Berger M. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is down-regulated in cystic fibrosis. Am J Respir Cell Mol Biol. 1995;13:257–61. doi: 10.1165/ajrcmb.13.3.7544594. [DOI] [PubMed] [Google Scholar]
- 42.Calahan MD, Lewis Rs. Regulation of chloride channels in lymphocytes. Curr Topic Membr. 1994;42:103–29. [Google Scholar]
- 43.Moss RB, Hsu YP, Olds L. Reduced IL-10 secretion by T cells expressing mutant CFTR is mimicked by inhibition of Cl− flux [Abstr.] Am J Resp Crit Care Med. 1997;155:A198. [Google Scholar]
- 44.Alton EWF, Stern M, Farley R, et al. Cationic lipid-mediated CFTR gene transfer to the lungs and nose of patients with cystic fibrosis: a double-blind placebo-controlled trial. Lancet. 1999;353:947–54. doi: 10.1016/s0140-6736(98)06532-5. [DOI] [PubMed] [Google Scholar]
- 45.Wagner JA, Messner AH, Moran ML, et al. A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in CF patients with antrostomies [Abstr.] Pediatr Pulmonol. 1999;19(Suppl.):223–4. doi: 10.1089/104303402760128577. [DOI] [PubMed] [Google Scholar]