

Abstract
We examined the role of autoantibodies to β2-GPI and prothrombin (PT) in the inhibition of annexin V binding to cardiolipin (CL) and the association with clinical manifestations of the anti-phospholipid syndrome (APS). Plasma samples from 59 patients with anti-phospholipid (aPL) antibodies were studied. Affinity purification of total IgG and IgG anti-ß2-GPI antibodies was performed using staphylococcal protein A and phospholipid liposomes. Annexin V binding to CL was significantly inhibited by 31/59 (53%) aPL+ plasma samples. There was a significant association between annexin V inhibition and elevated levels of IgG anti-cardiolipin (aCL) (r = −0·62; P < 0·001), IgG anti-ß2-GPI (r = −0·67; P < 0·001) and a weaker association with lupus anti-coagulant (r = −0·27; P = 0·05). There was no association with other isotypes of aCL and anti-ß2-GPI or with anti-PT of any isotype. In patients with clinical manifestations of the APS there were higher levels of IgG aCL (median (range) Z score): 10·0 (0–17·6) versus 5·0 (0–16·1); P = 0·03), IgG anti-ß2-GPI (4·5 (0–11·3) versus 0·9 (0–9·7); P = 0·02) and greater inhibition of annexin V binding to CL (−3·4 (−11·4–0·6) versus−1·1 (−10·8–1·2); P = 0·22). Odds ratios for the laboratory assays and the presence of clinical manifestations of the APS varied between 0·38 and 4·16, with the highest values for IgG aCL (4·16), IgG anti-ß2-GPI (3·28) and annexin V inhibition (2·85). Additional experiments with affinity-purified IgG antibodies indicated that inhibition of annexin V binding was dependent upon the concentration of ß2-GPI and anti-ß2-GPI antibodies. These results indicate that inhibition of annexin V binding to procoagulant phospholipid surfaces is dependent upon anti-ß2-GPI antibodies and suggest a role for annexin V in the pathogenesis of the APS.
Keywords: annexin V, anti-phospholipid antibodies, β2-glycoprotein I
Introduction
Anti-phospholipid (aPL) antibodies are a heterogeneous group of circulating autoantibodies directed against negatively charged phospholipids and protein ‘cofactors’[1–3]. There are at least two distinct groups of aPL antibodies. Type I occur predominantly in patients with infections such as syphilis, infectious mononucleosis and AIDS. These bind directly to phospholipid in vitro and have no clinical sequelae [4–7]. Type II are frequently found in patients with autoimmune diseases such as systemic lupus erythematosus (SLE). In vitro they bind to serum proteins such as β2-GPI and prothrombin (PT) which associate with negatively charged phospholipids such as cardiolipin (CL) through charge interactions [8–11]. These antibodies are implicated in the pathogenesis of the thrombotic events which characterize the anti-phospholipid syndrome (APS) [12–21].
The precise pathogenic mechanisms underlying the APS are still unknown. A variety of in vitro effects have been attributed to autoimmune aPL antibodies, including endothelial cell activation [22–24], platelet activation [25–27] and modulation of coagulation mechanisms leading to acquired protein C resistance [28]. Recent studies have suggested that inhibition of annexin V binding to procoagulant surfaces may be an additional mechanism through which aPL antibodies mediate their in vivo pathogenic effects [29,30]. The aim of the present study was to examine the role of autoantibodies to β2-GPI and PT, the two most common antigenic targets of autoimmune aPL antibodies, in this phenomenon and the association with clinical manifestations of the APS.
PATIENTS and METHODS
Patients
Fifty-nine patients with aPL antibodies, determined by ELISA (IgG anti-cardiolipin (aCL)) or functional coagulation assays (lupus anti-coagulant), identified through the Lupus Clinic or service laboratories at the Queen Elizabeth II Health Sciences Centre were included in the study. Clinical diagnoses were determined retrospectively based upon clinical assessment supported by appropriate diagnostic techniques (computed tomography, ultrasound and venography of the lower limbs, echocardiography). Twenty-nine (49%) patients had one or more of the core manifestations of the APS [18], namely venous or arterial thrombosis and recurrent (≥ 2) fetal loss. Nine of these 29 patients also fulfilled the American College of Rheumatology criteria for SLE [31]. An additional 18 patients had SLE without clinical manifestations of the APS and four patients had aPL antibodies without SLE or the APS. To determine the potential effect of anti-coagulation on inhibition of annexin V binding to CL, plasma samples were examined from 20 patients receiving heparin (median (range) partial thromboplastin time (PTT): 88·4 s (32·3–150·0 s)). These were collected during the post-operative period following cardiac bypass surgery. Plasma was also collected from another 20 patients attending an anti-coagulation clinic and taking warfarin for a variety of venous and arterial thrombotic disorders (median (range) INR: 2·5 (2–4)). Control plasma samples were collected from 14 healthy individuals. Peripheral venous blood was collected in sodium citrate tubes, centrifuged at 3000 rev/min for 30 min and the plasma stored at −70°C until use.
Purification of aPL antibodies
Phospholipid liposomes were used for purification of aPL antibodies as previously described by others [9,32]. In brief, CL:phosphatidylcholine:cholesterol liposomes were prepared in a ratio of 5:20:8 by evaporation under a stream of nitrogen. Dried lipids were resuspended in plasma, maintaining the final concentration of CL at 3 mg/ml, and incubated for 1 h at 37°C. After diluting 1:5 in 25 mm TBS pH 7·4, liposomes and bound material were pelleted by centrifugation at 23 000 g for 25 min and washed twice in TBS. The liposomal pellet was dissolved in 2% n-octyl-b-d-glucopyranoside. IgG was isolated from the bound material in the dissolved pellet and from the unbound material by affinity chromatography with staphylococcal Protein A. Purified proteins were dialysed and concentrated by ultrafiltration and quantified by capture ELISA as previously described [33].
ELISA for autoantibodies to CL (standard and direct)
Linbro 96-well EIA microtitre plates (ICN Biomedicals, Costa Mesa, CA) were coated with 45 μg/ml cardiolipin (Sigma, St Louis, MO) in 95% ethanol, 30 μl/well and incubated overnight at 4°C uncovered in order for ethanol to evaporate. The plates were post-coated with 10% fetal calf serum (FCS; Gibco, Gaithersberg, MD) in 0·02 m Tris-buffered saline (TBS) pH 7·4 for 2 h at room temperature. After three washes with TBS–Tween, 100 μl of plasma samples diluted with 10% FCS in TBS–Tween (1:100 for IgG, 1:50 for IgM and IgA) were added to duplicate wells and incubated for 1 h at room temperature. The plates were washed again and 100 μl of alkaline phosphatase-conjugated goat anti-human IgG, IgM, or IgA (Sigma) diluted 1:1000 in TBS–Tween containing 10% FCS were added to each well. After incubating at room temperature for 1 h, plates were washed three times and colour developed in the dark using p-nitrophenyl phosphate (Sigma) in diethanolamine buffer pH 9·8, 100 μl/well. The reaction was expressed in optical density (OD) units read with a Molecular Devices Emax microplate reader at 405 nm. The results were expressed in Z values [34] calculated using the OD results from 10 normal controls on each plate. A positive result was defined as a Z score of ≥ 2 (i.e. ≥ 2 s.d. above the mean of normals).
A modified ELISA was used for the detection of direct binding to CL by affinity-purified IgG fractions at a uniform concentration of 20 μg/ml. The essential difference was the exclusion of ß2-GPI and other ‘cofactors’ from the assay by the use of 0·3% gelatin to postcoat the wells and in the diluents. In addition, any non-specific binding of antibody to buffer-coated wells was subtracted from the OD in the CL-coated wells. The results were expressed in Z values and a positive result was defined as a Z score of ≥ 2.
Purification of ß2-GPI
β2-GPI was purified from normal human pooled plasma using cardiolipin-affinity and ion-exchange chromatography as previously described [35]. The purity of the protein was confirmed by SDS–PAGE using a wide range of molecular weight markers (6·5–205 kD; Sigma) with both coomassie brilliant blue and silver staining. The protein concentration was determined by UV spectrometry at 280 nm using the ß2-GPI extinction coefficient of 0·94.
ELISA for anti-ß2-GPI antibodies
Half of the wells in a Corning gamma-irradiated 96-well EIA microtitre plate (Fisher Scientific, Nepean, ON) were coated with 10 μg/ml of ß2-GPI in carbonate coating buffer pH 9·6, 50 μl/well. The other half of the plate was coated with carbonate buffer only. Plates were covered and incubated overnight at 4°C. After three washes in 0·02 m TBS, plates were postcoated with 1% bovine serum albumin (BSA) in TBS, 100 μl/well for 1 h at room temperature. The plates were washed three times in TBS–Tween, and 100 μl of plasma diluted with 1% BSA in TBS–Tween (1:100 for IgG, 1:50 for IgM and IgA) were incubated in duplicate wells on both the ß2-GPI-coated and buffer-coated sides of the plates concurrently for 3 h at room temperature. The plates were washed again in TBS–Tween and 100 μl alkaline phosphatase-conjugated goat anti-human IgG, IgM, or IgA diluted 1:1000 in TBS–Tween containing 1% BSA were added to each well. After incubating for 1·5 h at room temperature, plates were washed and the reaction developed in the dark with p-nitrophenyl phosphate in diethanolamine buffer pH 9·8, 100 μl/well. The OD was read with a Molecular Devices Emax microplate reader at 405 nm. The OD values of the buffer-coated wells were subtracted from the OD values of the ß2-GPI-coated wells, and Z values calculated using plasma samples from 10 normal controls on each plate. A positive result was defined as a Z score of ≥ 2.
ELISA for anti-PT antibodies
This protocol was identical to that for the detection of anti-ß2-GPI antibodies except that the wells were coated with 10 μg/ml PT (Enzyme Research Labs, South Bend, IN) in carbonate coating buffer, 50 μl/well. A positive result was defined as a Z score of ≥ 2.
Identification of lupus anti-coagulant
Screening for the detection of lupus anti-coagulant (LA) was determined by prolongation of either the activated partial thromboplastin time (aPTT) or the dilute Russell viper venom time (dRVVT). Confirmation of an antibody inhibitor was demonstrated by failure to correct with normal plasma and normalization by exogenous phospholipid using the LAC Confirm reagent according to the manufacturer's instructions (Instrumentation Laboratory, Lexington, MA). Prolongation of in vitro coagulation by 20% or more (LAC ratio > 1·2) by test plasma compared with normal plasma or exogenous phospholipid was used to indicate the presence of a lupus anti-coagulant. This methodology is in keeping with international recommendations for the detection of LA [36].
Competitive ELISA for annexin V binding to CL
Linbro 96-well EIA microtitre plates (ICN Biomedicals) were coated with 45 μg/ml CL (Sigma) in 95% ethanol, 30 μl/well, and incubated overnight at 4°C uncovered in order for ethanol to evaporate. The plates were postcoated with 100 μl/well of annexin V buffer (20 mm HEPES, 132 mm NaCl, 6 mm KCl, 2·5 mm CaCl2·2H2O, 1 mm MgSO4·7H2O, 5 mm d-glucose, 0·5% BSA) pH 7·4 for 1 h at room temperature. After one wash with annexin V buffer, biotin-conjugated annexin V (Cedarlane, Fort Washington, PA) in annexin V buffer was diluted 1:1 with plasma samples and added to plates in duplicate wells, 100 μl/well for 1 h at room temperature. The final concentration of annexin V in the wells was 1 μg/ml. Plates were washed three times with annexin V buffer and 100 μl of alkaline phosphatase-conjugated Extravidin (Sigma) diluted 1:5000 in annexin V buffer were added to each well. After incubating at room temperature for 1 h, plates were washed three times and colour developed in the dark using p-nitrophenyl phosphate (Sigma) in diethanolamine buffer pH 9·8, 100 μl/well. The reaction was expressed in OD units read with a Molecular Devices EMax microplate reader at 405 nm. Results were expressed as Z values calculated using the OD results from 10 normal controls on each plate. A significant result was defined as a Z score of −2·0 or less (i.e. two or more s.d. below the mean of normal controls), indicating inhibition of annexin V binding to CL by test samples compared with controls. On each plate the binding of annexin V to CL in the absence of patient or control plasma was confirmed.
Affinity-purified IgG antibodies were also tested for their ability to inhibit annexin V binding to CL. The protocol was identical except that 100 μg/ml IgG were added to wells with 1 μg/ml biotin-conjugated annexin V in annexin V buffer, with and without 10 μg/ml of ß2-GPI (final concentrations in wells), 50 μl/well for 1 h at room temperature. The amount of annexin V inhibition was quantified by OD, where the lowest OD indicated the greatest inhibition. In some experiments the results were expressed as Z values calculated from the difference between the OD for the same IgG sample with and without ß2-GPI. In this case the difference between ODs increases with the amount of annexin V inhibition. Thus a significant result was defined as a Z value of + 2·0 or more (i.e. two or more s.d. above the mean of normal controls), indicating inhibition of annexin V binding to CL by affinity-purified IgG from test samples compared with IgG from controls.
Statistical analysis
Differences between groups were examined by Mann–Whitney U-test. The association between groups was examined by Pearson correlation coefficient.
Results
Anti-phospholipid antibodies and inhibition of annexin V binding by plasma samples
The prevalence of aCL, anti-ß2-GPI, anti-PT antibodies and LA activity, and the antibody levels are summarized in Table 1. IgG aCL and LA were present in 73% and 82% of plasma samples, respectively, while both were detected in 59% of patients.
Table 1.
Autoantibody profile of 59 anti-phospholipid (aPL)+ plasma samples
| Z score > 2 (no. of samples) | Z score (median (range)) | |
|---|---|---|
| aCL | ||
| IgG | 43 (73%) | 6·4 (0–17·6) |
| IgM | 41 (70%) | 3·9 (0–76·2) |
| IgA | 25 (42%) | 1·3 (0–30·3) |
| Anti-β2-GPI | ||
| IgG | 30 (51%) | 2·0 (0–11·3) |
| IgM | 33 (56%) | 3·3 (0–46·9) |
| IgA | 41 (70%) | 8·3 (0–239·2) |
| Anti-PT | ||
| IgG | 28 (48%) | 1·0 (0–84·0) |
| IgM | 23 (39%) | 1·3 (0–9·2) |
| IgA | 23 (39%) | 1·3 (0–11·0) |
| LA (ratio > 1·2) | 46/56 (82%) | 1·5 (0·9–2·8) |
| Annexin V inhibition | 31 (53%) | −2·2 (1·2 −11·4) |
aCL, Anti-cardiolipin; PT, prothrombin; LA, lupus anti-coagulant.
Significant inhibition of annexin V binding to CL was induced by 53% of plasma samples (Table 1). The coefficient of variation for intra-assay variability was 8% and for interassay variability was 20%. There was a significant association between annexin V inhibition and elevated levels of IgG aCL (r = −0·62; P < 0·001), IgG anti-ß2-GPI (r = −0·67; P < 0·001) and a weaker association with LA (r = −0·27; P = 0·05) (Table 2, Fig. 1). There was no association with other isotypes of aCL and anti-ß2-GPI or with anti-PT of any isotype (Table 2, Fig. 1).
Table 2.
Association between autoantibody specificity and inhibition of annexin V binding to cardiolipin (CL) in 59 anti-phospholipid (aPL)+ plasma samples
| Autoantibody specificity | r | P |
|---|---|---|
| IgG aCL | −0·62 | < 0·001 |
| IgG anti-β2-GPI | −0·67 | < 0·001 |
| IgG anti-PT | 0·19 | 0·15 |
| IgM aCL | −0·11 | 0·4 |
| IgM anti-β2-GPI | −0·1 | 0·43 |
| IgM anti-PT | 0·15 | 0·27 |
| IgA aCL | −0·01 | 0·92 |
| IgA anti-β2-GPI | −0·07 | 0·59 |
| IgA anti-PT | 0·24 | 0·07 |
| LA | 0·27 | 0·05 |
PT, Prothrombin.
Fig. 1.
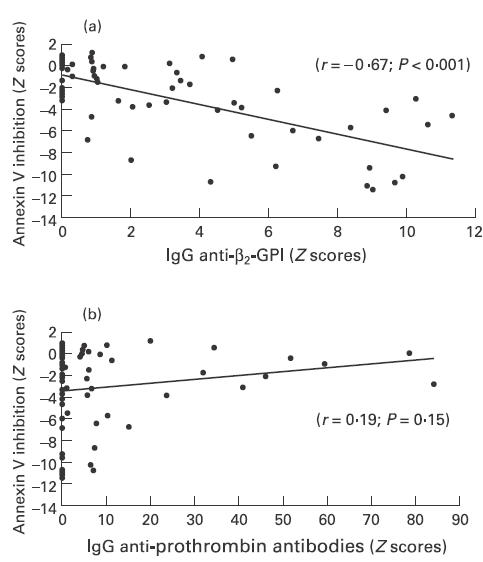
Correlation between inhibition of annexin V binding to cardiolipin, IgG anti-ß2-GPI (a) and IgG anti-prothrombin antibodies (b) in 59 anti-phospholipid antibody-positive plasma samples.
To determine the potential effect of anti-coagulation on inhibition of annexin V binding to CL, plasma samples were examined from 20 patients receiving heparin and from another 20 patients taking warfarin. Heparin therapy significantly inhibited annexin V binding to CL (Z score < −2·0) in 12/20 (60%) samples. The median (range) Z score was −3·4 (3·1–10·1). In contrast, warfarin therapy significantly inhibited annexin V binding in only 2/20 (10%) samples with a median (range) Z score of −1·2 (0·8–5·0).
Association between clinical manifestations of the APS and laboratory findings
In patients with clinical manifestations of the APS there was greater inhibition of annexin V binding to CL (median (range) Z score: −3·4 (−11·4 to 0·6) versus−1·1 (−10·8 to 1·2); P = 0·22) and significantly higher levels of IgG aCL (median (range) Z score: 10·0 (0–17·6) versus 5·0 (0–16·1); P = 0·03) and IgG anti-ß2-GPI (median (range) Z score: 4·5 (0–11·3) versus 0·9 (0–9·7); P = 0·02) (Table 3). Odds ratios for the presence of the APS varied between 0·38 and 4·16 with the highest values for IgG aCL (4·16), IgG anti-ß2-GPI (3·28) and annexin V inhibition (2·85).
Table 3.
Association between anti-phospholipid syndrome* (APS), serologic abnormalities and inhibition of annexin V binding
| APS present (n = 29) | APS absent (n = 30) | P | Odds ratio (95% confidence intervals) | |
|---|---|---|---|---|
| IgG aCL† | 10·0 (0–17·6) | 5·0 (0–16·1) | 0·03 | 4·17 (1·15–15·04) |
| IgG anti-β2-GPI | 4·5 (0–11·3) | 0·9 (0–9·7) | 0·02 | 3·28 (1·13–9·54) |
| IgG anti-PT | 0·7 (0–78·5) | 4·4 (0–84·0) | 0·61 | 0·62 (0·22–1·73) |
| IgM aCL | 5·6 (0·4–76·2) | 3·8 (0–63·6) | 0·47 | 0·95 (0·31–2·89) |
| IgM anti-β2-GPI | 3·6 (0–46·9) | 2·2 (0–43·8) | 0·59 | 1·64 (0·58–4·62) |
| IgM anti-PT | 1·5 (0–9·2) | 0·8 (0–9·1) | 0·62 | 0·92 (0·32–2·61) |
| IgA aCL | 1·4 (0–30·3) | 1·2 (0–24·6) | 0·96 | 0·92 (0·33–2·59) |
| IgA anti-β2-GPI | 10·3 (0–239·2) | 7·9 (0–110·4) | 0·87 | 0·69 (0·23–2·11) |
| IgA anti-PT | 1·3 (0–9·5) | 2·1 (0–11·0) | 0·22 | 0·38 (0·13–1·13) |
| LA | 1·7 (1–2·8) | 1·5 (0·9–2·7) | 0·20 | 2·78 (0·64–12·10) |
| Annexin V inhibition | −3·4 (−11·4–0·6) | −1·1 (−10·8–1·2) | 0·22 | 2·85 (0·99–8·21) |
APS, Anti-phospholipid syndrome characterized clinically by either venous thrombosis, arterial thrombosis or recurrent (≥ 2) fetal loss.
All values (median (range)) are Z scores with the exception of lupus anti-coagulant (LA) activity, which is expressed as a ratio.
PT, Prothrombin.
Affinity-purified anti-ß2GPI antibodies and inhibition of annexin V binding
IgG was isolated from 10 plasma samples known to have high levels of anti-ß2-GPI antibodies, using phospholipid liposomes and staphylococcal Protein A affinity chromatography. All liposomal eluates had high levels of anti-ß2-GPI antibody activity (median (range) Z score: 12·9 (11·2–14·5)) when examined at a concentration of 5 μg/ml of total IgG. In contrast, IgG isolated from the unbound material following incubation with liposomes (supernatants) was substantially depleted of anti-ß2-GPI activity (median (range) Z score: 3·1 (0–10·6)) when examined at the same antibody concentration.
Inhibition of annexin V binding to CL by total IgG, IgG anti-ß2-GPI-enriched (eluates) and IgG anti-ß2-GPI-depleted (supernatants) samples was examined at a uniform antibody concentration of 100 μg/ml in the presence of ß2-GPI 10 μg/ml. Six of the 10 IgG samples from liposomal eluates demonstrated significant inhibition of annexin V binding (Z score > 2), which in all cases was greater than that seen with comparable amounts of total IgG and IgG from liposomal supernatants (Fig. 2). Levels of anti-ß2-GPI were significantly higher in the six liposomal eluates which inhibited annexin V binding compared with the four that did not (median (range) Z score: 13·6 (12·4–14·5) versus 11·7 (11·2–12·7); P = 0·02). Inhibition was dependent upon the concentration of ß2-GPI (Fig. 3a) and there was also a clear dose–response effect between the concentration of anti-ß2-GPI antibody and inhibition of annexin V binding (Fig. 3b). Increasing the concentration of total IgG to 1400 µg/ml caused a significant inhibition of annexin V binding in only two of the 10 samples with Z scores of 2·2 and 2·1. Finally, IgG isolated from liposomal supernatants with antibody binding to PT (n = 3; median (range) Z score: 7·5 (2·6–12·30)) or CL (direct) (n = 4; median (range) Z score: 7·6 (5·0–12·7)) in the absence of anti-ß2-GPI antibody activity (all Z scores < 2) did not inhibit annexin V binding to CL (data not shown).
Fig. 2.
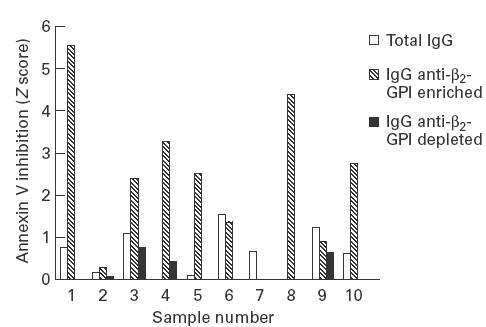
Comparison of inhibition of annexin V binding to cardiolipin by 100 μg/ml of total IgG, IgG anti-ß2-GPI-enriched and IgG anti-ß2-GPI-depleted antibodies isolated from 10 patients with known autoimmune aPL antibodies.
Fig. 3.
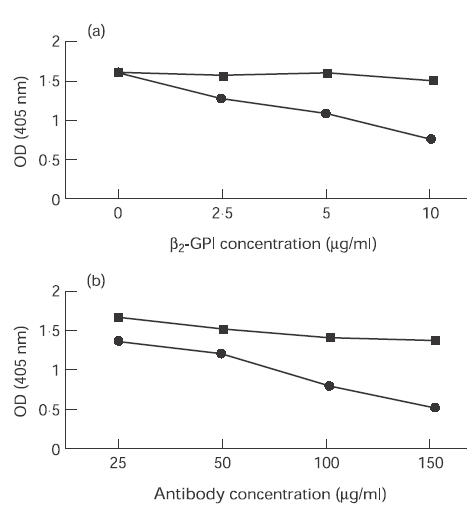
(a) The effect of increasing concentrations of ß2-GPI from 0 to 10 μg/ml on annexin V binding to cardiolipin (CL) while maintaining the concentration of affinity-purified IgG anti-ß2-GPI antibodies at 100 μg/ml. (b) The effect of increasing concentrations of affinity-purified IgG anti-ß2-GPI antibodies from 25 to 150 μg/ml on annexin V binding to CL while maintaining the concentration of ß2-GPI at 10 μg/ml. The results in (a) and (b) are from experiments using antibodies isolated from a single representative patient (•) and control (▪). OD, Optical density.
Discussion
Autoimmune aPL antibodies are detected either by solid-phase binding assays or functional phospholipid-dependent coagulation assays. They comprise a heterogeneous group of autoantibodies which bind in vitro not to phospholipids but predominantly to plasma proteins (‘cofactors’) which associate with negatively charged phospholipids through charge interactions. The most frequent targets of this autoantibody response are ß2-GPI [8–11] and PT [37,38]. Although these antibodies are linked to the clinical manifestations of the APS [12–21], in particular venous and arterial thrombosis, such events occur in only 50–60% of cases as defined by conventional assays for antibody detection.
The precise pathogenic mechanisms which are responsible for the clinical manifestations of the APS are still unknown. The results of in vitro experiments have suggested endothelial cell activation [22–24], platelet activation [25–27] and modulation of coagulation mechanisms such as acquired protein C resistance [28] as potential candidates. Rand et al. [29] have recently proposed inhibition of annexin V binding to procoagulant surfaces as an additional mechanism. They demonstrated that the amount of annexin V eluted from the surface of cultured human umbilical vein endothelial cells and trophoblasts was significantly reduced by preincubation with total IgG isolated from three patients with APS. This was associated with a concurrent increase in the procoagulant effect of similarly treated cells. Subsequent studies [30] with non-cellular phospholipid-coated surfaces demonstrated comparable inhibition of annexin V binding by total IgG in the presence of ß2-GPI and plasma samples from a limited number of patients with APS.
The results of the present study support and expand the findings of Rand et al. [29,30]. A competitive ELISA-based assay indicated that 53% of plasma samples from 59 patients with documented aPL antibodies caused significant inhibition of annexin V binding to CL-coated plates. This was strongly correlated with plasma levels of anti-ß2-GPI antibodies. Additional experiments with total IgG and highly enriched anti-ß2-GPI antibody preparations derived from six patients with aPL antibodies confirmed this association. Failure to demonstrate a similar effect by affinity-purified antibodies from four additional patients may have been due in part to lower antibody levels, although other antibody characteristics such as isotype subclass or affinity and epitope specificity for ß2-GPI may also play an important role in producing this in vitro effect. Previous studies by Pierangeli et al. [39] have demonstrated similar competitive in vitro binding for CL between annexin V and IgG from patients with APS in the presence of ß2-GPI. The previous studies by Rand et al. [29,30] used unfractionated preparations of total IgG and thus could not determine which autoantibody specificity was responsible for the observed effects. The results of the present study indicate that autoantibody reactivity directed against ß2-GPI, rather than PT or CL (direct), is primarily responsible for inhibition of annexin V binding.
The pathogenic significance of these in vitro findings is unclear. Annexin V belongs to a family of ubiquitous, non-glycosylated proteins which bind to phospholipids in the presence of calcium [40]. It is found in the placenta, a number of cell types including endothelial cells, and is also present in the circulation [41,42]. Annexin V has been shown to bind preferentially to negatively charged phospholipids either immobilized on solid surfaces or expressed in biologic membranes [43,44]. Its ability to displace phospholipid-dependent coagulation factors and replace them with clusters of annexin V molecules may account for its reported in vitro and in vivo anti-coagulant effects. Rand et al. [29,30] have postulated that displacement or inhibition of annexin V binding to procoagulant surfaces on endothelial cells, platelets and trophoblasts may significantly impair this physiologic anti-coagulant function and account for some of the clinical manifestations of the APS. Additional work is required to confirm these in vitro findings and to determine their in vivo significance in patients with APS.
The results of the current study suggest that inhibition of annexin V binding may be a valuable predictor of risk for the development of clinical manifestations of the APS, an issue that was not addressed in the initial studies by Rand et al. [29,30]. Although we examined the cumulative risk for clinical disease we did not examine the temporal association between clinical events and changes in annexin V inhibition. This would be of considerable interest in view of the recent report that levels of anti-ß2-GPI antibodies fall significantly around the time of a thrombotic event in patients with APS [45]. Although the assay is artefactually modified by plasma samples from patients who are anti-coagulated with therapeutic doses of heparin, it is largely unaffected by concurrent warfarin therapy.
Additional laboratory studies are required to clarify the role of annexin V in the aetiology of the APS and its interaction with other proposed pathogenic mechanisms. Prospective clinical studies should also examine the temporal relationship between abnormalities in serologic and functional assays of aPL antibodies and the varied clinical manifestations of the syndrome.
Acknowledgments
Supported by grants from the Medical Research Council and the Arthritis Society of Canada. J.G.H. is a Clinical Associate of the Arthritis Society of Canada.
REFERENCES
- 1.McNeil HP, Chesterman CN, Krilis Sa. Immunology and clinical importance of antiphospholipid antibodies. Adv Immunol. 1991;49:193–280. doi: 10.1016/s0065-2776(08)60777-4. [DOI] [PubMed] [Google Scholar]
- 2.Roubey Ras. Autoantibodies to phospholipid-binding plasma proteins: a new view of lupus anticoagulants and other ‘antiphospholipid’ autoantibodies. Blood. 1994;84:2854–67. [PubMed] [Google Scholar]
- 3.Roubey R. Immunology of the antiphospholipid syndrome. Arthritis Rheum. 1996;39:1444–54. doi: 10.1002/art.1780390903. [DOI] [PubMed] [Google Scholar]
- 4.Harris EN, Gharavi AE, Wasley GD, Hughes Grv. Use of an enzyme-linked immunosorbent assay and of inhibition studies to distinguish between antibodies to cardiolipin from patients with syphilis or autoimmune disorders. J Infect Dis. 1988;157:23–31. doi: 10.1093/infdis/157.1.23. [DOI] [PubMed] [Google Scholar]
- 5.Hunt JE, McNeil HP, Morgan GJ, Crameri RM, Krilis Sa. A phospholipid-β2-glycoprotein I complex is an antigen for anticardiolipin antibodies occurring in autoimmune disease but not with infection. Lupus. 1992;1:75–81. doi: 10.1177/096120339200100204. [DOI] [PubMed] [Google Scholar]
- 6.Vaarala O, Palosuo T, Kleemola M, et al. Anticardiolipin response in acute infections. Clin Immunol Immunopathol. 1986;41:8–15. doi: 10.1016/0090-1229(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 7.Zarrabi MH, Zucker S, Miller F, et al. Immunologic and coagulation disorders in chlorpromazine-treated patients. Ann Intern Med. 1979;91:194–9. doi: 10.7326/0003-4819-91-2-194. [DOI] [PubMed] [Google Scholar]
- 8.McNeil HP, Simpson RJ, Chesterman CN, Krilis Sa. Antiphospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: β2-glycoprotein I (apolipoprotein H) Proc Natl Acad Sci USA. 1990;87:4120–5. doi: 10.1073/pnas.87.11.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galli M, Comfurius P, Maassen C, et al. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet. 1990;335:1544–7. doi: 10.1016/0140-6736(90)91374-j. [DOI] [PubMed] [Google Scholar]
- 10.Bevers EM, Galli M. Beta 2-glycoprotein I for binding of anticardiolipin antibodies to cardiolipin. Lancet. 1990;336:952–3. doi: 10.1016/0140-6736(90)92330-k. [DOI] [PubMed] [Google Scholar]
- 11.Matsuura E, Igarashi Y, Fujimoto M, Ichikawa K, Koike T. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet. 1990;366:177. doi: 10.1016/0140-6736(90)91697-9. [DOI] [PubMed] [Google Scholar]
- 12.Asherson R, Khamashta M, Ordi-Ros J, et al. The ‘primary’ antiphospholipid syndrome: major clinical and serologic features. Medicine. 1988;68:366–74. [PubMed] [Google Scholar]
- 13.Love PE, Santoro Sa. Antiphospholipid antibodies: anticardiolipin and lupus anticoagulant in systemic lupus erythematosus (SLE) and non-SLE disorders. Ann Intern Med. 1990;112:682–98. doi: 10.7326/0003-4819-112-9-682. [DOI] [PubMed] [Google Scholar]
- 14.Cines DB McCrae. The antiphospholipid-protein syndrome. J Clin Immunol. 1995;15(Suppl.):86S–100S. doi: 10.1007/BF01540898. [DOI] [PubMed] [Google Scholar]
- 15.Shapiro Ss. The lupus anticoagulant/antiphospholipid syndrome. Annu Rev Med. 1996;47:533–53. doi: 10.1146/annurev.med.47.1.533. [DOI] [PubMed] [Google Scholar]
- 16.Nahass Gt. Antiphospholipid antibodies and the antiphospholipid syndrome. J Am Acad Dermatol. 1997;36:149–68. doi: 10.1016/s0190-9622(97)70274-3. [DOI] [PubMed] [Google Scholar]
- 17.Alarcon-Segovia D, Sanchez-Guerrero J. Primary antiphospholipid syndrome. J Rheumatol. 1989;16:482–8. [PubMed] [Google Scholar]
- 18.Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome. Arthritis Rheum. 1999;42:1309–11. doi: 10.1002/1529-0131(199907)42:7<1309::AID-ANR1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 19.Alarcon-Segovia D, Deleze M, Oria CV, et al. Antiphospholipid antibodies and the antiphospholipid syndrome in systemic lupus erythematosus: a prospective analysis of 500 consecutive cases. Medicine. 1989;68:353–65. doi: 10.1097/00005792-198911000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Ginsberg JS, Wells PS, Brill-Edwards P, et al. Antiphospholipid antibodies and venous thromboembolism. Blood. 1995;86:3685–91. [PubMed] [Google Scholar]
- 21.Simioni P, Prandoni P, Zanon E, et al. Deep venous thrombosis and lupus anticoagulant. A case-control study. Thromb Haemost. 1996;76:187–9. [PubMed] [Google Scholar]
- 22.Del Papa N, Meroni PL, Tincani A, et al. Relationship between anti-phospholipid and anti-endothelial cell antibodies: further characterization of the reactivity on resting and cytokine activated endothelial cell. Clin Exp Rheumatol. 1992;10:37–42. [PubMed] [Google Scholar]
- 23.Simantov R, LaSala JM, Lo SK, et al. Activation of cultured vascular endothelial cells by antiphospholipid antibodies. J Clin Invest. 1995;96:2211–9. doi: 10.1172/JCI118276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Del Papa N, Guidali L, Sala A, et al. Endothelial cells as target for antiphospholipid antibodies: human polyclonal and monoclonal anti-β2-glycoprotein I antibodies react in vitro with endothelial cells through adherent β2-glycoprotein I and induce endothelial activation. Arthritis Rheum. 1997;40:551–61. doi: 10.1002/art.1780400322. [DOI] [PubMed] [Google Scholar]
- 25.Asano T, Furie BC, Furie B. Platelet binding properties of monoclonal lupus autoantibodies produced by human hybridomas. Blood. 1985;66:1254–60. [PubMed] [Google Scholar]
- 26.Rauch J, Meng Q-H, Tannenbaum H. Lupus anticoagulant and antiplatelet properties of human hybridoma autoantibodies. J Immunol. 1987;139:2598–604. [PubMed] [Google Scholar]
- 27.Khamashta MA, Harris EN, Gharavi AE, et al. Immune mediated mechanism for thrombosis: antiphospholipid antibody binding to platelet membranes. Ann Rheum Dis. 1988;47:849–54. doi: 10.1136/ard.47.10.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galli M, Ruggeri L, Barbui T. Differential effects of anti-β2-glycoprotein-I and antiprothrombin antibodies on the anticoagulant activity of activated protein C. Blood. 1998;91:1999–2004. [PubMed] [Google Scholar]
- 29.Rand JH, Wu X-X, Andree HAM, Lockwood CJ, Guller S, Scher J, Harpel Pc. Pregnancy loss in the antiphospholipid antibody syndrome—a possible thrombogenic mechanism. N Engl J Med. 1997;337:154–60. doi: 10.1056/NEJM199707173370303. [DOI] [PubMed] [Google Scholar]
- 30.Rand JH, Wu X-X, Andree HAM, Ross JBA, Rusinova E, Gascon-Lema MG, Calandri C, Harpel Pc. Antiphospholipid antibodies accelerate plasma coagulation by inhibiting annexin-V binding to phospholipids: a ‘lupus procoagulant’ phenomenon. Blood. 1998;92:1652–60. [PubMed] [Google Scholar]
- 31.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 32.Pengo V, Thiagarajan P, Shapiro SS, Heine Mj. Immunological specificity and mechanism of action of IgG lupus anticoagulants. Blood. 1987;70:69–76. [PubMed] [Google Scholar]
- 33.Hanly JG, Hong C, Issekutz A. Beta 2-glycoprotein I and anticardiolipin antibody binding to resting and activated cultured endothelial cells. J Rheumatol. 1996;23:1543–9. [PubMed] [Google Scholar]
- 34.Hanly JG, Hong C, Smith S, Fisk Jd. A prospective analysis of cognitive function and anticardiolipin antibodies in systemic lupus erythematosus. Arthritis Rheum. 1999;42:728–34. doi: 10.1002/1529-0131(199904)42:4<728::AID-ANR16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 35.McNeil HP, Krilis SA, Chesterman Cn. Purification of antiphospholipid antibodies using a new affinity method. Thromb Res. 1988;52:641–8. doi: 10.1016/0049-3848(88)90136-3. [DOI] [PubMed] [Google Scholar]
- 36.Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. Thromb Haemostasis. 1995;74:1185–90. [PubMed] [Google Scholar]
- 37.Loeliger A. Prothrombin as cofactor in the circulating anticoagulant in systemic lupus erythematosus? Thromb Diath Haemorrh. 1959;3:237. [Google Scholar]
- 38.Bevers EM, Galli M, Barbui T, Comfurius P, Zwaal Rfa. Lupus anticoagulant IgGs (LA) are not linked to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb Haemost. 1991;66:629–32. [PubMed] [Google Scholar]
- 39.Pierangeli SS, Dean J, Goldsmith GH, Branch DW, Gharavi A, Harris En. Studies on the interaction of placental anticoagulant protein I, ß2-glycoprotein I, and antiphospholipid antibodies in the prothrombinase reaction and in the solid phase anticardiolipin assays. J Lab Clin Med. 1996;128:194–201. doi: 10.1016/s0022-2143(96)90011-6. [DOI] [PubMed] [Google Scholar]
- 40.Romisch J, Paques Ep. Annexins: calcium-binding proteins of multi-functional importance? Med Microbiol Immunol. 1991;180:109–19. doi: 10.1007/BF00206115. [DOI] [PubMed] [Google Scholar]
- 41.Romisch J, Schuller E, Bastian B, Burger T, Dunkel FG, Schwinn A, Hartmann AA, Paques Ep. Annexin I-VI. Quantitative determination in different human cell types and in plasma after myocardial infarction. Blood Coag Fibrin. 1992;3:11–19. [PubMed] [Google Scholar]
- 42.Flaherty MJ, West S, Heimark R, Fujikawa K, Tait Fj. Placental anticoagulant protein I. Measurement in extracellular fluids and cells of the hemostatic system. J Lab Clin Med. 1990;115:174–83. [PubMed] [Google Scholar]
- 43.Andree HAM, Stuart MCA, Hermens WTH, Reutelingsperger CPM, Hemker HC, Frederik PM, Willems Gm. Clustering of lipid-bound annexin V may explain its anticoagulant effect. J Biol Chem. 1992;267:17907–13. [PubMed] [Google Scholar]
- 44.van Heerde WL, Poort S, van't Veer C, Reutelingsperger CPM, de Groot Pg. Binding of recombinant annexin V to endothelial cells: effect of annexin V binding on endothelial-cell-mediated thrombin formation. Biochem J. 1994;302:305–11. doi: 10.1042/bj3020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomez-Pacheco L, Villa AR, Drenkard C, Cabiedes J, Cabral AR, Alarcon-Segovia D. Serum anti-β2-glycoprotein-I and anticardiolipin antibodies during thrombosis in systemic lupus erythematosus. Am J Med. 1999;106:417–23. doi: 10.1016/s0002-9343(99)00053-4. [DOI] [PubMed] [Google Scholar]